MPX Quality Control
This tutorial details the first steps of data analysis to quality control and clean up the MPX data output from Pixelator.
After completing this tutorial, you should be able to:
- Use the molecule rank plot to manually set cell calling thresholds and filter low-quality cells.
- Aggregate data across samples and visualize sample-level QC metrics like number of cells.
- Check distributions of quality metrics like molecule counts and graph connectivity.
- Identify and remove cell outliers using the antibody count distribution metric Tau.
Setup
First, we will load packages necessary for downstream processing.
from pathlib import Path
from pixelator import read
from pixelator.plot import molecule_rank_plot, cell_count_plot, scatter_umi_per_upia_vs_tau
import seaborn as sns
sns.set_style("whitegrid")
DATA_DIR = Path("<path to the directory to save datasets to>")
Load data
In this tutorial we will continue where we left off after the previous tutorial Data handling and load the combined object directly. As described previously, this merged object contains four samples: a resting and PHA stimulated PBMC sample, both in duplicate. Note that if you continue directly from the previous tutorial, you don’t need to load the data again.
pg_data_combined_pxl_object = read(DATA_DIR/ "combined_data.pxl")
# In this tutorial we will mostly work with the AnnData object
# so we will start by selecting that from the pixel data object
pg_data_combined = pg_data_combined_pxl_object.adata
Cell calling: Edge rank plot
Here, we use the molecule rank plot to perform an additional quality control of the called cells, to make a manual adjustment to the number of cells that were called by Pixelator. This removes cells that deviate from the component size distribution, and might not represent whole cells.
molecule_rank_df = pg_data_combined.obs[["sample", "molecules"]].copy()
molecule_rank_df["rank"] = molecule_rank_df.groupby(["sample"])["molecules"].rank(
ascending=False, method="first"
)
fig, ax = molecule_rank_plot(molecule_rank_df, group_by="sample")
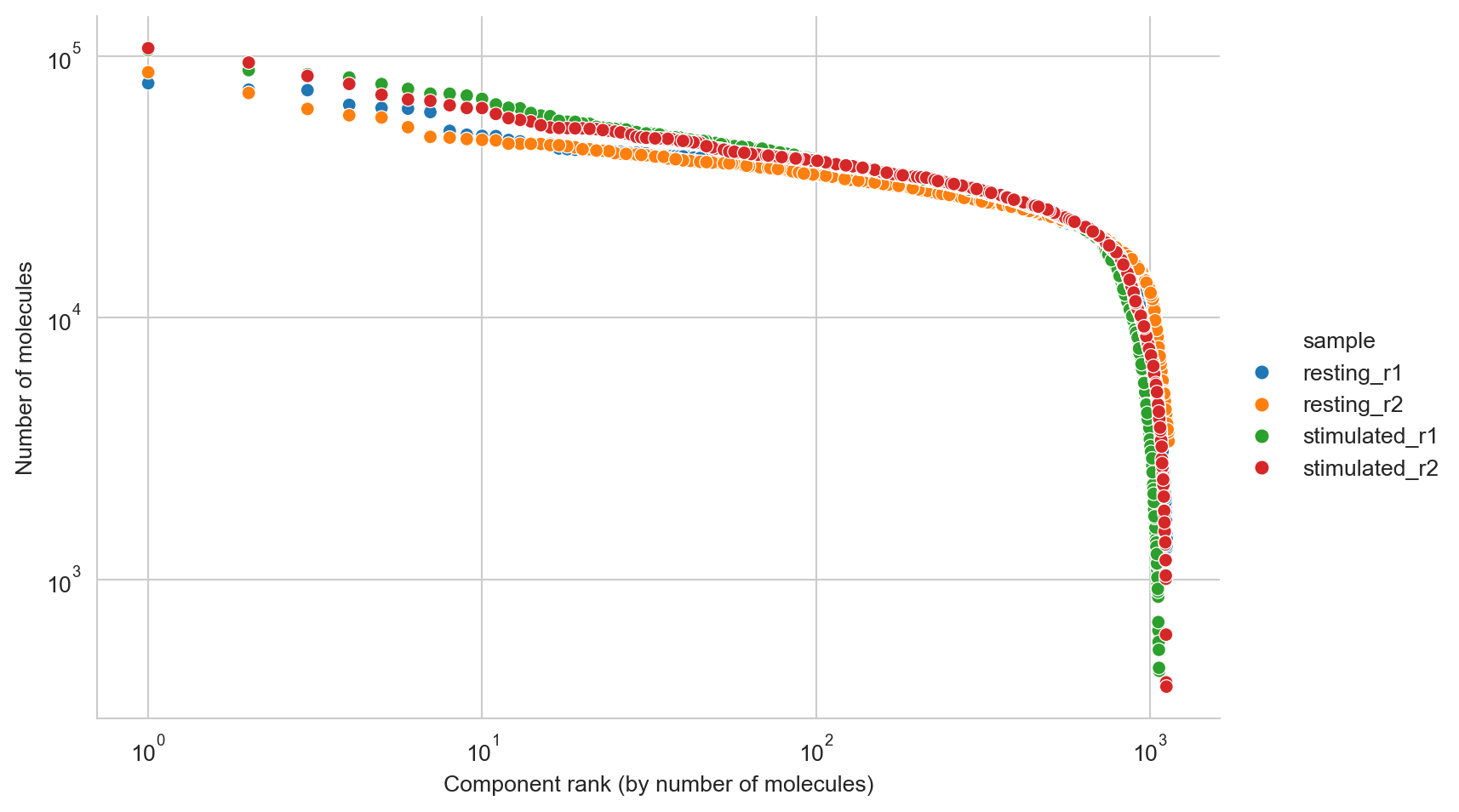
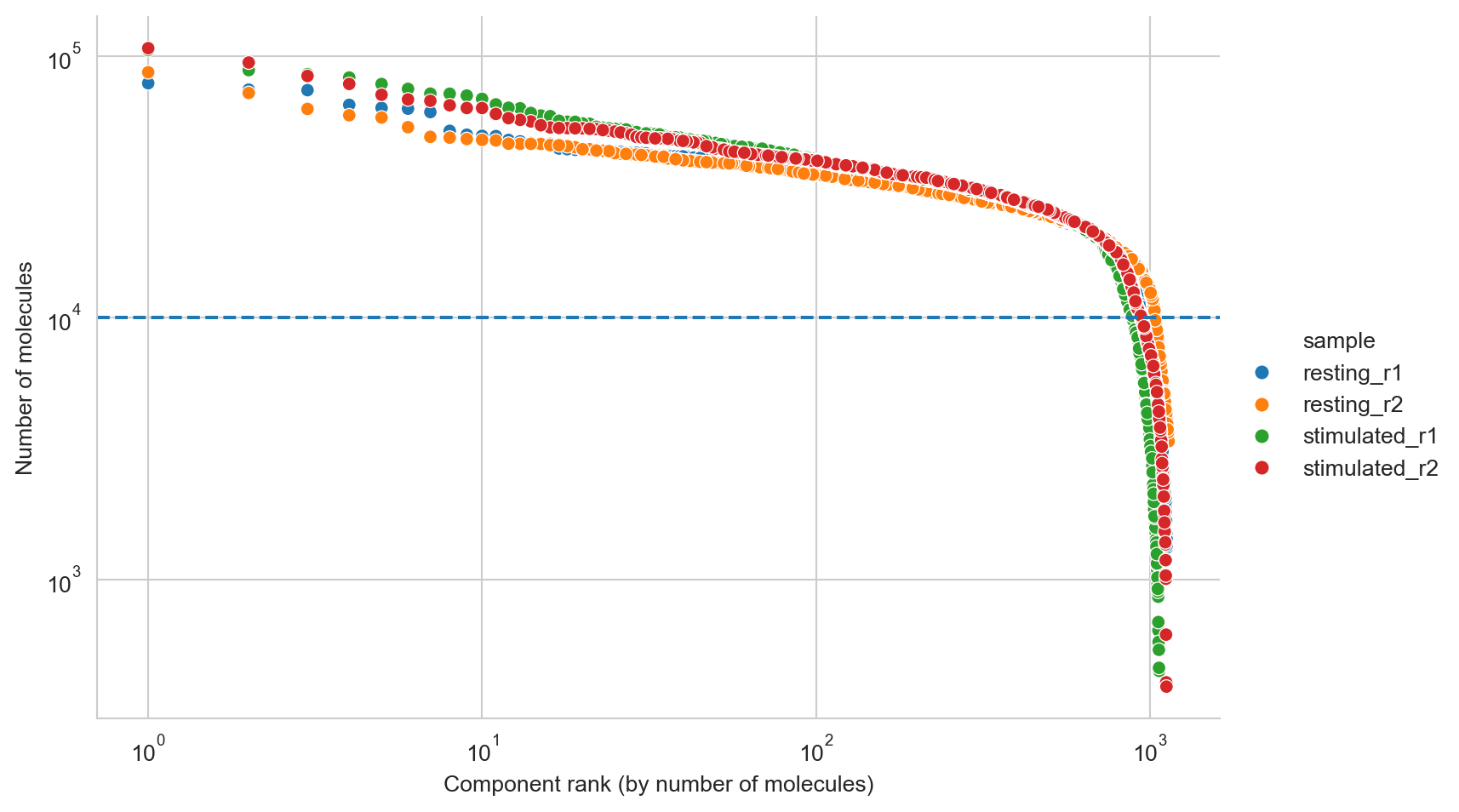
It looks like components are declining rapidly in size at around 10000 molecules, and we will thus set a manual cutoff at that point, represented by a dashed line.
fig, ax = molecule_rank_plot(molecule_rank_df, group_by="sample")
ax.axhline(10000, linestyle="--")
# Filter cells to have at least 10000 edges
pg_data_combined = pg_data_combined[pg_data_combined.obs["molecules"] >= 10000]
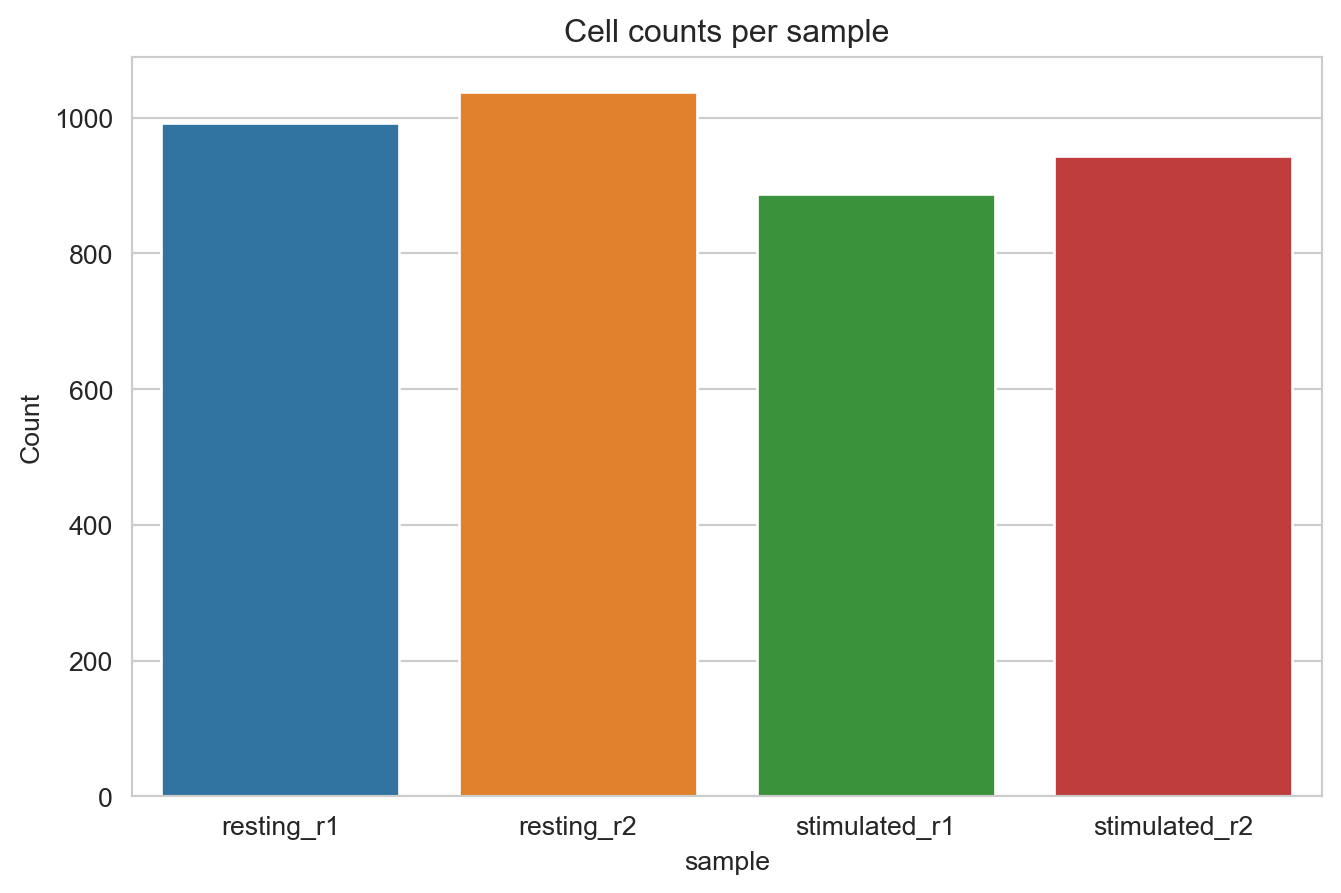
Here, we plot the number of called cells per condition and replicate.
cells_per_sample_df = (
pg_data_combined.obs.groupby("sample").size().to_frame(name="size").reset_index()
)
fig, ax = cell_count_plot(pg_data_combined.obs, color_by="sample")
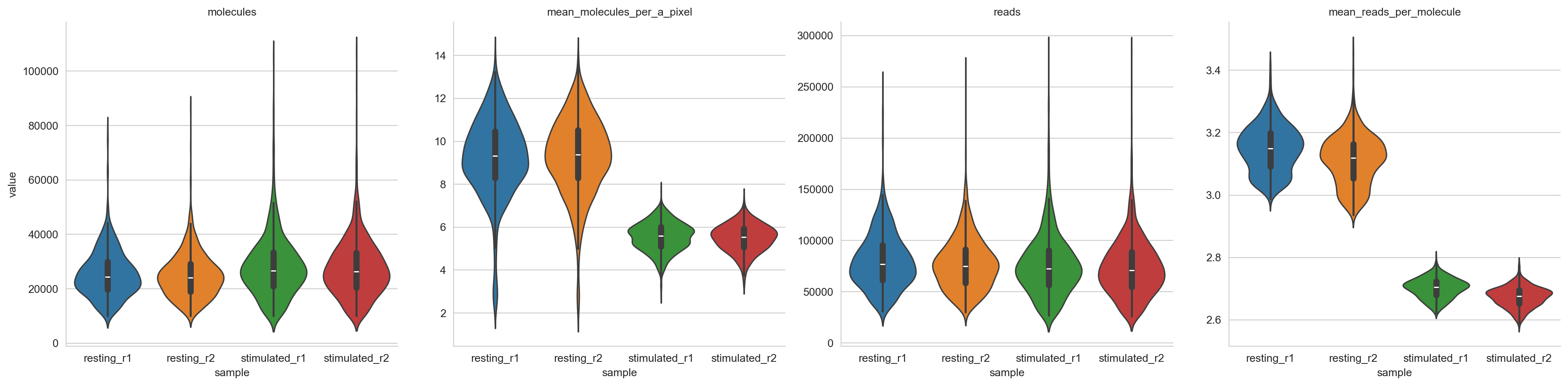
Here, we visualize the distribution of some metrics among components.
metrics_per_sample_df = pg_data_combined.obs[
["sample", "molecules", "mean_molecules_per_a_pixel", "reads", "mean_reads_per_molecule"]
].melt(id_vars=["sample"])
metrics_plot = sns.catplot(
data=metrics_per_sample_df,
x="sample",
col="variable",
y="value",
kind="violin",
sharex=True,
sharey=False,
margin_titles=True,
hue="sample",
).set_titles(col_template="{col_name}")
metrics_plot
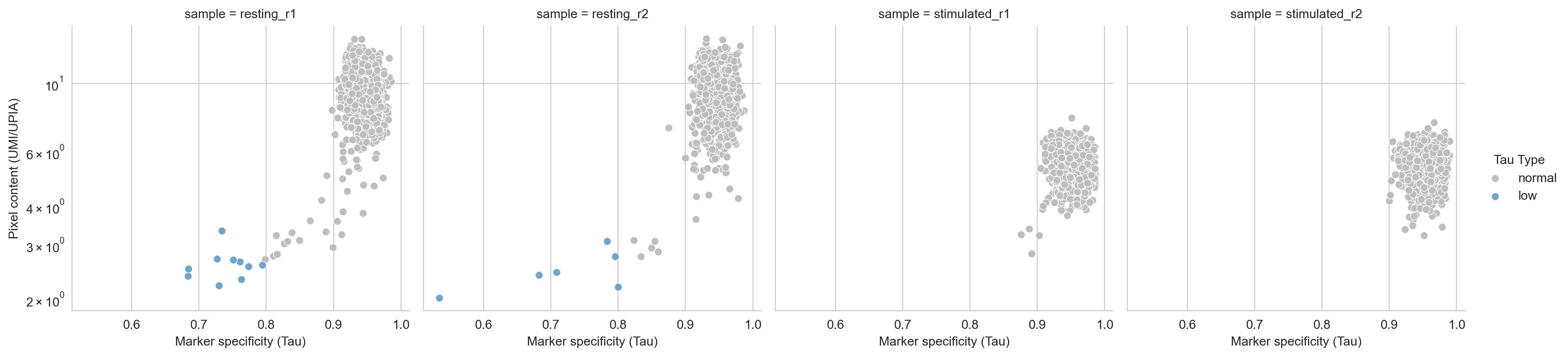
Antibody count distribution outlier removal
Here, we have plotted the umi_per_upia stat, reflecting the mean
molecules per DNA-pixel A, against
Tau,
and colored each component by Pixelator’s classification of Tau
(low, normal, or high). It looks like Pixelator has accurately
picked out some outliers that might be an outlier antibody complex or a
component that has low specificity, binding many more different types of
antibodies than we would expect from a normal cell. As these outliers
are likely a technical artefact, we will remove them from the analysis.
tau_metrics_df = pg_data_combined.obs[["sample", "tau", "mean_molecules_per_a_pixel", "tau_type"]]
tau_metrics_df = tau_metrics_df.rename(columns={"mean_molecules_per_a_pixel": "umi_per_upia"})
fig, ax = scatter_umi_per_upia_vs_tau(tau_metrics_df, group_by="sample")
Going through the above steps we have performed critical quality control by filtering low-quality cells and identifying outliers. With a clean, high-quality MPX dataset in hand, we are ready to proceed to the next step, in which we will be leveraging protein abundance for the annotation of different cell populations.
If you want to pause here and save the filtered data for the next tutorial, you can do so with the following code.
components_to_keep = pg_data_combined_pxl_object.adata[
(pg_data_combined_pxl_object.adata.obs["molecules"] >= 10000)
& (pg_data_combined_pxl_object.adata.obs["tau_type"] == "normal")
].obs.index
pg_data_combined_pxl_object.filter(components=components_to_keep).save(DATA_DIR/ "combined_data_filtered.pxl", force_overwrite=True)