Patch analysis
This tutorial will introduce patch analysis, a methodology encompassing various analytical tasks that extend patch detection in PNA data. Patch detection is a technique designed to identify localized regions, or “patches,” formed by one cell type on another. Within the framework of PNA data, a patch is precisely characterized as a connected subgraph exhibiting enrichment for specific protein markers that are not present in other areas of the graph. Depending on the biological context, these patches can signify diverse entities, including fragments of other cells, entire distinct cells (such as platelets), or cellular debris.
For instance, patch analysis holds significant relevance in co-culture experiments where cell-cell interactions are prominent. Patch detection aids in segmenting these interacting cells, thereby providing valuable insights into their interplay. Core aspects of patch analysis involve quantifying patches and examining their protein composition.
After completing this tutorial, you will be able to:
-
Understand the prerequisites for patch analysis.
-
Identify protein markers for patch detection.
-
Run patch detection.
-
Examine the protein composition of patches.
Patch analysis is an experimental feature in pixelatorR and might
change in the near future. Use it with caution and please provide
feedback to the developers if you encounter any issues or have
suggestions for improvements.
Setup
library(pixelatorR)
library(SeuratObject)
library(dplyr)
library(tidyr)
library(stringr)
library(ggplot2)
library(here)
library(patchwork)
library(harmony)
library(tidygraph)
library(tibble)
library(Seurat)
plot_colors <-
c(
"#866CCD", "#4A73C0", "#4AAF9D", "#5EA857",
"#CB73A9", "#DB5D61", "#D07B5B", "#E6BB43",
"#A28EDB", "#6D92D1", "#75C5B5"
)
BASEURL <- "https://pixelgen-technologies-datasets.s3.eu-north-1.amazonaws.com/pna-datasets/v1"
files_to_download <-
c("PNA065_CARTcells_Raji_24hrs_1to1_S05_S5.layout.pxl", "PNA065_CARTcells_S01_S1.layout.pxl",
"PNA065_Raji_S04_S4.analysis.pxl", "PNA065_Raji_S04_S4.layout.pxl",
"PNA066_CARTcells_Raji_24hrs_1to1_S05_S5.layout.pxl", "PNA066_CARTcells_S01_S1.layout.pxl",
"PNA066_Raji_S04_S4.analysis.pxl", "PNA066_Raji_S04_S4.layout.pxl")
for (f in files_to_download) {
if (!file.exists(file.path(DATA_DIR, f))) {
download.file(paste0(BASEURL, "/", f), destfile = file.path(DATA_DIR, f), method = "curl")
}
}
Load data
In this tutorial we will use a dataset containing chimeric antigen receptor (CAR) T-cells co-cultured for 24 hours with a cancerous B-cell line (Raji) and additionally two control datasets with pure CAR T-cells and Raji populations that were not co-cultured.
These particular CAR T-cells are engineered to express a chimeric antigen receptor targeting the CD19 antigen on Raji cells which allows the T-cells to recognize and eliminate the malignant Raji cells. This type of engineered immune cells have emerged as a transformative approach to cancer immunotherapy but are still in a relatively early stage of development. Challenges such as antigen escape, T cell exhaustion and limited efficacy in solid tumors remain and highlight the need for a deeper understanding of the mechanisms of action of CAR T-cells. It is however becoming increasingly clear that the spatial organization of the membrane-bound proteins and their ability to form clusters and complexes on the surface of the engineered T-cells are key factors in their activation, function and persistence.
Besides the spatial organization of the membrane-bound proteins, which we will not explore in this tutorial, this dataset is also a prime example of how to use PNA data to study a process called trogocytosis. Trogocytosis is a form of cell-to-cell interaction where one cell transfer fragments (or “patches”) of its cell membrane to another cell. In the CAR T-cell context this means that the CAR T-cells incorporate fragments of the Raji cell membrane into their own cell membrane. This can have important functional consequences for the CAR T-cells, such as tumor escape due to reduced antigen density on the target cell, increased T-cell exhaustion and even fratricide among the CAR T-cells. Until now there has not been a technology to study this process at scale and looking at this many targets simultaneously, so we are excited to add patch detection to your analysis toolbox for these types of experiments and datasets. To start exploring the CAR T-cell dataset begin by downloading and loading the data as described below.
DATA_DIR <- "path/to/local/folder"
Sys.setenv("DATA_DIR" = DATA_DIR)
# The folders the data is located in:
data_files <- c(
coculture1 = file.path(DATA_DIR, "PNA065_CARTcells_Raji_24hrs_1to1_S05_S5.layout.pxl"),
coculture2 = file.path(DATA_DIR, "PNA066_CARTcells_Raji_24hrs_1to1_S05_S5.layout.pxl"),
cart1 = file.path(DATA_DIR, "PNA065_CARTcells_S01_S1.layout.pxl"),
cart2 = file.path(DATA_DIR, "PNA066_CARTcells_S01_S1.layout.pxl"),
raji1 = file.path(DATA_DIR, "PNA065_Raji_S04_S4.layout.pxl"),
raji2 = file.path(DATA_DIR, "PNA066_Raji_S04_S4.layout.pxl")
)
# Read PXL files as a list of Seurat objects
obj_list <- lapply(data_files, ReadPNA_Seurat)
# Combine files into a single Seurat object
pg_data_combined <- merge(obj_list[[1]], y = obj_list[-1], add.cell.ids = names(obj_list))
# Add meta data describing the sample and donor origin
pg_data_combined <- AddMetaData(
pg_data_combined,
metadata = data.frame(id = colnames(pg_data_combined)) %>%
mutate(condition = str_remove(id, "[1|2]_.*"),
rep = str_extract(id, "[1|2]") %>% as.integer()) %>%
column_to_rownames("id")
)
pg_data_combined
An object of class Seurat
159 features across 2948 samples within 1 assay
Active assay: PNA (159 features, 159 variable features)
6 layers present: counts.1, counts.2, counts.3, counts.4, counts.5, counts.6
Filter data
To ensure high data quality prior to patch detection, it’s essential to filter out low-quality cells. Our quality control criteria will include the number of UMIs, isotype fraction, and tau type. Detailed information regarding these quality control steps can be found in previous tutorials.
pg_data_combined <- pg_data_combined %>%
subset(tau_type == "normal") %>%
subset(isotype_fraction < 0.001) %>%
subset(n_umi >= 1e4)
Data-driven clustering
Dealing with co-culture data often means facing mixed cell abundance profiles across different conditions. To address this and simplify cell annotation, we’ll start by integrating our dataset using Harmony. Following integration, we will proceed with clustering and annotating the cells (refer to previous tutorials for a detailed guide on these initial steps if needed).
pg_data_combined <- pg_data_combined %>%
Normalize(method = "clr") %>%
ScaleData() %>%
RunPCA() %>%
RunHarmony(group.by.vars = "condition") %>%
RunUMAP(dims = 1:10, reduction = "harmony") %>%
FindNeighbors(dims = 1:10, reduction = "harmony") %>%
FindClusters(verbose = FALSE)
DimPlot(pg_data_combined, label = TRUE, label.size = 6, cols = plot_colors)
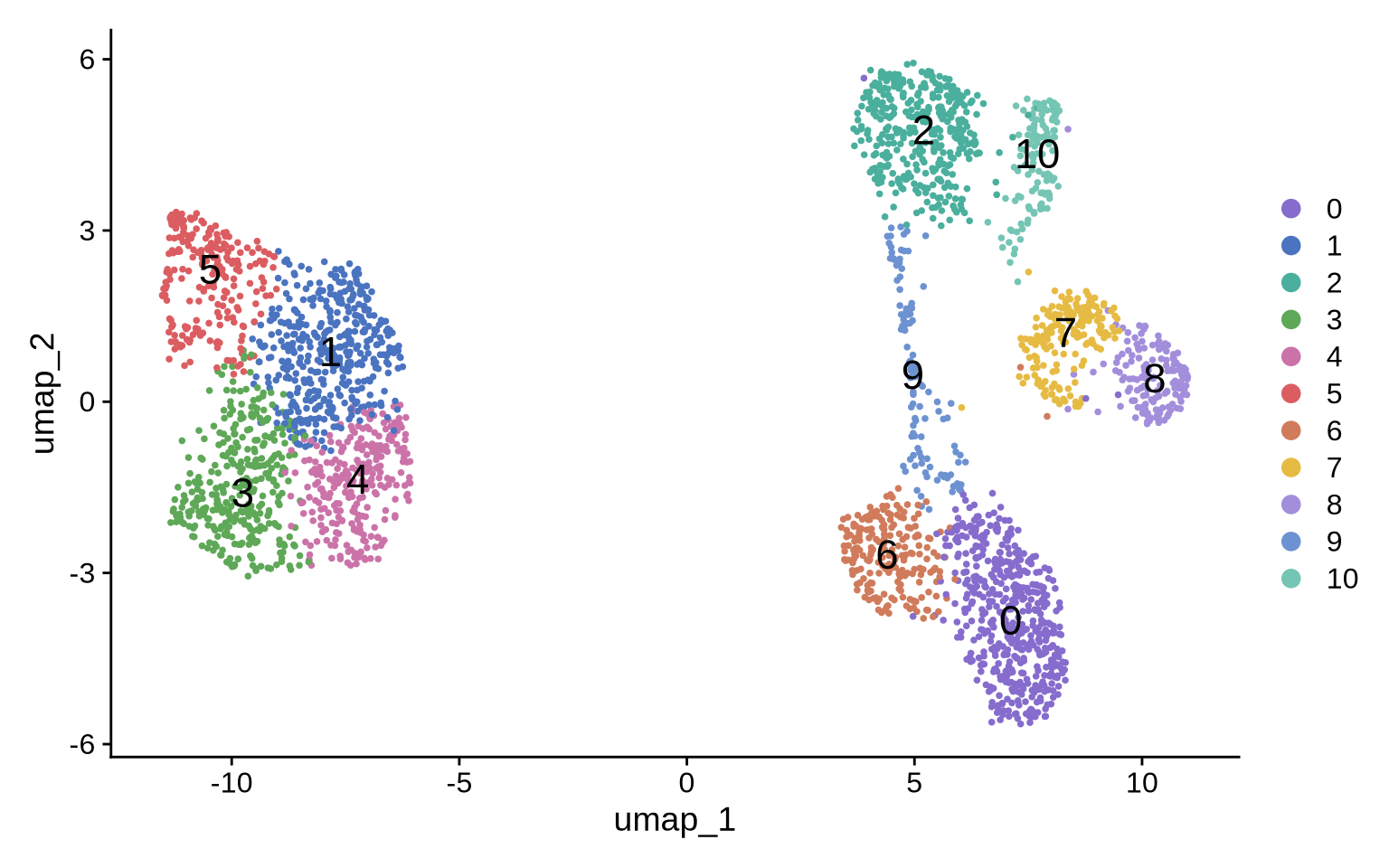
Annotation
Next, we run a differential test to identify cluster-specific protein markers. These markers will guide the annotation step. Here we simply classify clusters as “CD8T” if CD8 is differentially up-regulated, “CD4T” if CD4 is differentially up-regulated and “Raji” if CD21/CD19/CD22 are differentially up-regulated. Cell type annotations are saved to a new column called “celltype”.
Finally, we create a column combining the celltype annotation and condition.
de_markers <- FindAllMarkers(pg_data_combined, only.pos = TRUE, fc.name = "difference", mean.fxn = rowMeans)
de_markers %>%
group_by(cluster) %>%
arrange(-difference) %>%
slice_head(n = 10)
# A tibble: 110 × 7
# Groups: cluster [11]
p_val difference pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 3.11e-229 4.17 0.988 0.594 4.95e-227 0 CD8
2 2.77e-199 3.25 1 0.537 4.40e-197 0 CD162
3 9.61e- 85 3.08 1 0.66 1.53e- 82 0 CD44
4 1.35e- 91 2.70 1 0.646 2.15e- 89 0 CD3e
5 6.00e-110 2.66 1 0.549 9.53e-108 0 CD6
6 9.24e-153 2.66 1 0.658 1.47e-150 0 CD26
7 2.25e-168 2.61 1 0.506 3.58e-166 0 CD50
8 2.58e- 96 2.58 1 0.655 4.10e- 94 0 CD2
9 2.67e- 82 2.45 1 0.588 4.25e- 80 0 CD5
10 3.63e- 81 2.17 1 0.496 5.78e- 79 0 CD226
# ℹ 100 more rows
ann <- c(
"0" = "CD8T",
"1" = "Raji",
"2" = "CD8T",
"3" = "Raji",
"4" = "Raji",
"5" = "Raji",
"6" = "CD8T",
"7" = "CD4T",
"8" = "CD4T",
"9" = "CD4T",
"10" = "CD8T"
)
pg_data_combined$celltype <- ann[as.character(pg_data_combined$seurat_clusters)] %>% unname()
pg_data_combined$celltype_condition <-
paste(
pg_data_combined$celltype,
ifelse(
pg_data_combined$condition %in% c("cart", "raji"),
"alone",
"coculture"
),
sep = "_"
) %>% unname()
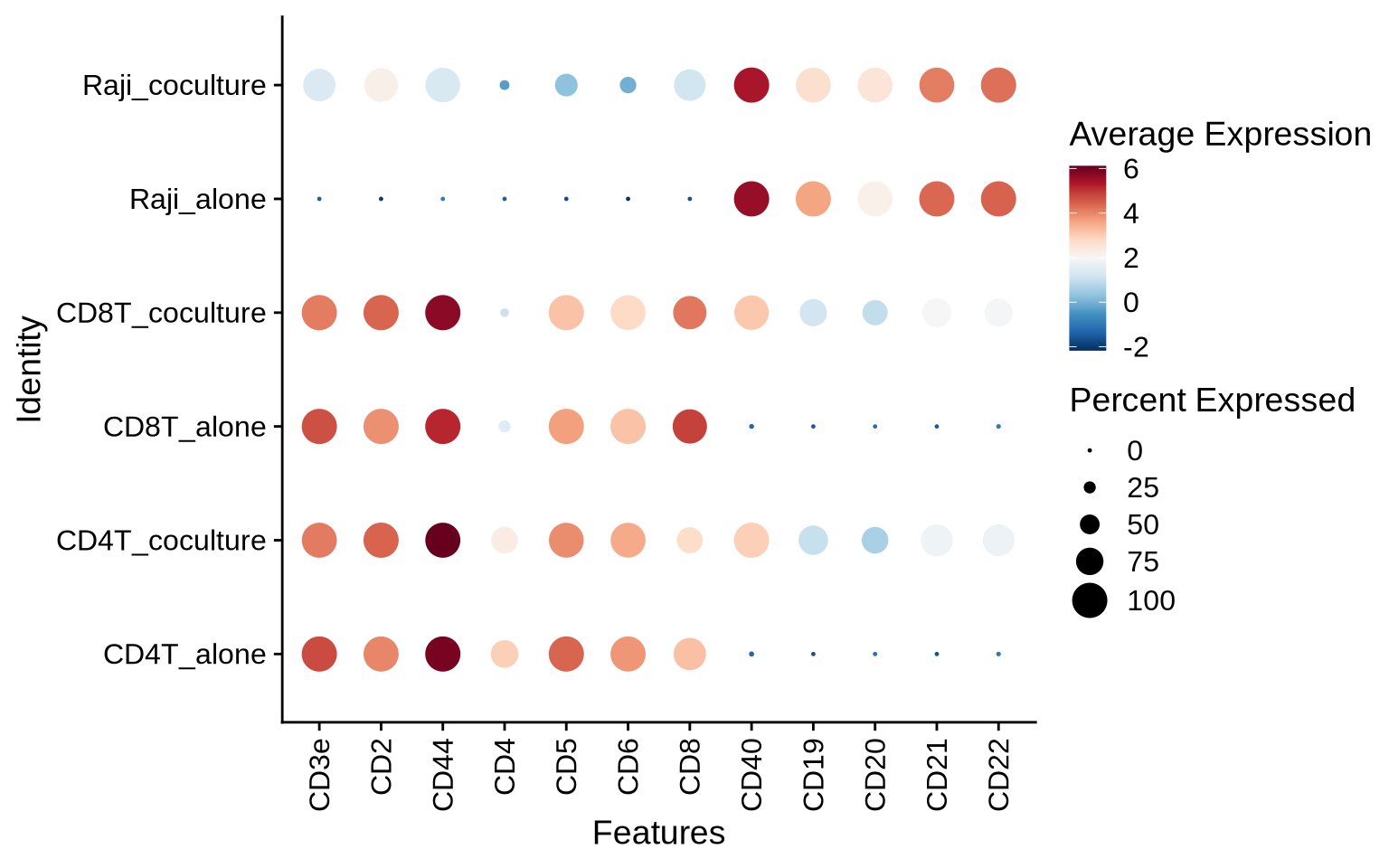
As a sanity check, we can visualize the abundance levels of selected T cell and Raji markers. As expected, the T cells from the co-culture experiment display elevated levels of Raji markers, indicating the presence of Raji patches on these cells. In the isolated conditions, the T cell populations are free of Raji markers and the Raji cells are free of T cell markers.
DotPlot(pg_data_combined, scale = FALSE,
features = c("CD3e", "CD2", "CD44", "CD4", "CD5", "CD6", "CD8", "CD40", "CD19", "CD20", "CD21", "CD22"),
group.by = "celltype_condition") &
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5)) &
scale_color_gradientn(colours = RColorBrewer::brewer.pal(n = 11, name = "RdBu") %>% rev())
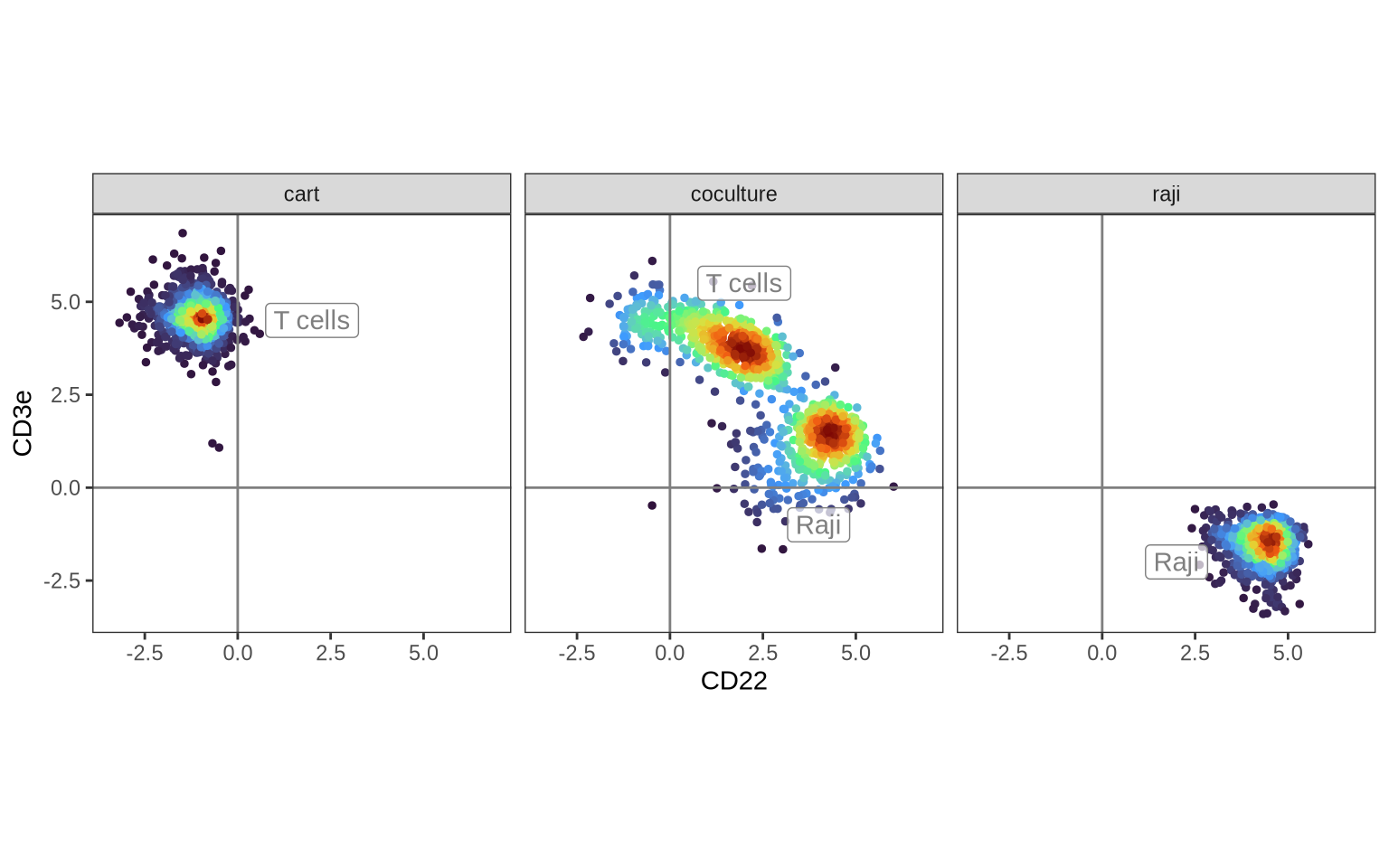
We can also plot a density scatter plot of a T cell (CD3e) and a Raji (CD22) marker protein to compare co-cultured cells with the isolated populations. Here, we clearly see that the T cell population gains CD22 in the co-culture condition. We also expect to see some bleedover from the T cells to the Raji cells, as evident by the slight increase in CD3e in the Raji co-culture population.
DensityScatterPlot(pg_data_combined, marker1 = "CD22", marker2 = "CD3e", facet_vars = "condition") &
geom_label(data = tibble(
marker1 = c(2, 2, 4, 2),
marker2 = c(4.5, 5.5, -1, -2),
label = c("T cells", "T cells", "Raji", "Raji"),
condition = c("cart", "coculture", "coculture", "raji"),
dens = NA_real_
), aes(marker1, marker2, label = label), alpha = 0.7) &
facet_grid(~condition) &
guides(color = "none")
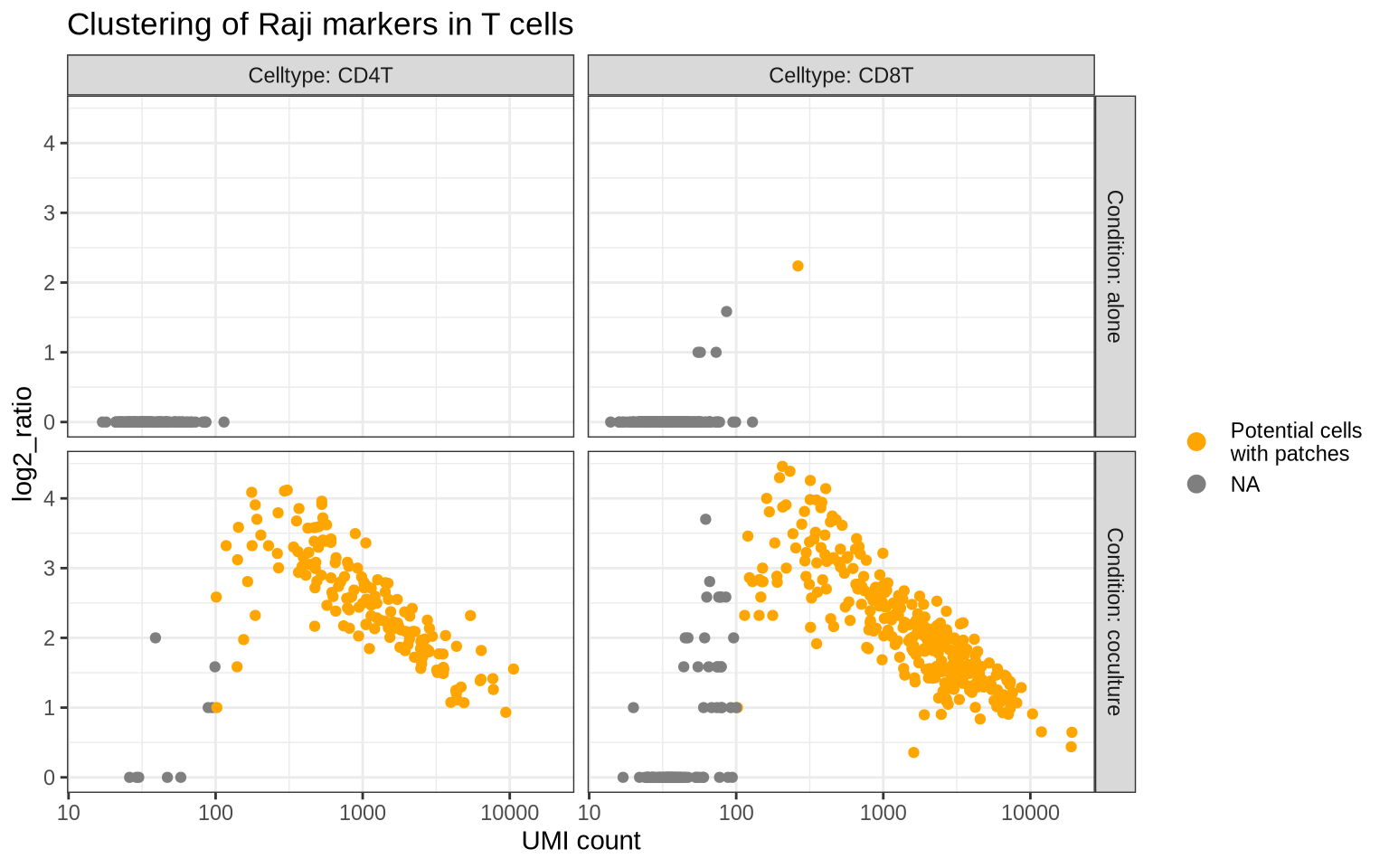
Patched cells detected by proximity scores
If the Raji patches on the T cells are large enough, it’s reasonable to expect elevated abundance levels for Raji protein markers on these T cells. We can also expect to see elevated proximity scores for these Raji markers since they should be aggregated in confined regions of the T cell graphs, thereby forming more connections with each other more than expected by random chance.
In the dot plot above, we saw that CD40, CD19, CD20, CD21 and CD22 are specific to Raji cells. We can compute a joint proximity score (log2_ratio) for these markers in the T cell populations to see if this expectation holds true.
The plot below shows the log2_ratio vs the total UMI count for the Raji markers in the T cells split by condition. As expected, virtually all T cells from the co-culture experiment have high log2_ratio values for the Raji markers, whereas the isolated T cell populations only display background levels.
This analysis is a good sanity check before proceeding to the patch detection step. Now we have support both from the abundance and proximity data that the T cells from the co-culture experiment are indeed patched with Raji cells.
raji_markers <- c("CD40", "CD19", "CD20", "CD21", "CD22")
counts <- FetchData(pg_data_combined, vars = raji_markers, layer = "counts")
counts <- tibble(component = rownames(counts), count = rowSums(counts))
# Compute joint log2_ratio for raji markers
prox_raji_markers <-
ProximityScores(pg_data_combined, meta_data_columns = "celltype_condition") %>%
filter(
celltype_condition %in% c(
"CD8T_alone",
"CD4T_alone",
"CD4T_coculture",
"CD8T_coculture"
)
) %>%
filter(marker_1 %in% raji_markers & marker_2 %in% raji_markers) %>%
group_by(celltype_condition, component) %>%
summarize(
join_count = sum(join_count),
join_count_expected_mean = sum(join_count_expected_mean),
.groups = "drop"
) %>%
collect() %>%
mutate(log2_ratio = log2(pmax(join_count, 1) / pmax(join_count_expected_mean, 1))) %>%
left_join(counts, by = c("component")) %>%
separate(celltype_condition,
into = c("celltype", "condition"),
sep = "_") %>%
mutate(label = case_when(
log2_ratio > 0.3 & count > 100 ~ "Potential cells\nwith patches",
TRUE ~ NA_character_
))
# Plot results
prox_raji_markers %>%
ggplot(aes(count, log2_ratio, color = label)) +
geom_point() +
scale_x_log10() +
theme_bw() +
facet_grid(paste0("Condition: ", condition) ~ paste0("Celltype: ", celltype)) +
labs(color = "") +
scale_color_manual(values = c("Potential cells\nwith patches" = "orange")) +
guides(color = guide_legend(override.aes = list(size = 3))) +
labs(x = "UMI count", title = "Clustering of Raji markers in T cells")
Patch detection
We will illustrate patch detection using the CD8 T-cell population from our co-culture experiment, where we anticipate finding Raji patches. A key prerequisite for patch detection is that we can only consider two populations at a time: a receiver population and a target population. In this example, our receiver population consists of CD8 T-cells, and the target population is the Raji cells. Note that we cannot detect patches from multiple cell types simultaneously. As previously observed, most CD8 T-cells in the co-culture experiment exhibit elevated levels of Raji markers, strongly suggesting they are patched with Raji cells. Crucially, the presence of the Raji population within the dataset is essential for identifying the specific protein markers that differentiate these two populations.
Before proceeding, we need to select population-specific protein markers – those that are highly specific and abundant for either CD8 T-cells or Raji cells. pixelatorR includes a convenient tool to help us easily identify these markers.
A key challenge here is that CD8 T-cells from the co-culture experiment show significant abundance levels of Raji marker proteins, complicating the identification of truly CD8T-cell-specific marker proteins. One might consider using pure CD8T-cell and Raji cell populations as a reference. However, such data may not always be accessible. More importantly, protein marker abundance levels can shift during co-culture, meaning that protein markers defined from isolated cells might not be optimal.
To tackle this, pixelatorR offers a method that unmixes abundance data to help identify “pure” abundance profiles for population marker identification. Let’s try it out. We’ll need to provide:
- A group column: such as “celltype_condition”
- A target population: for instance, “Raji_coculture”
- A receiever population: like “CD8T_coculture”
markers_df <- identify_markers_for_patch_analysis(
pg_data_combined,
group_by = "celltype_condition",
receiver_population = "CD8T_coculture",
target_population = "Raji_coculture"
)
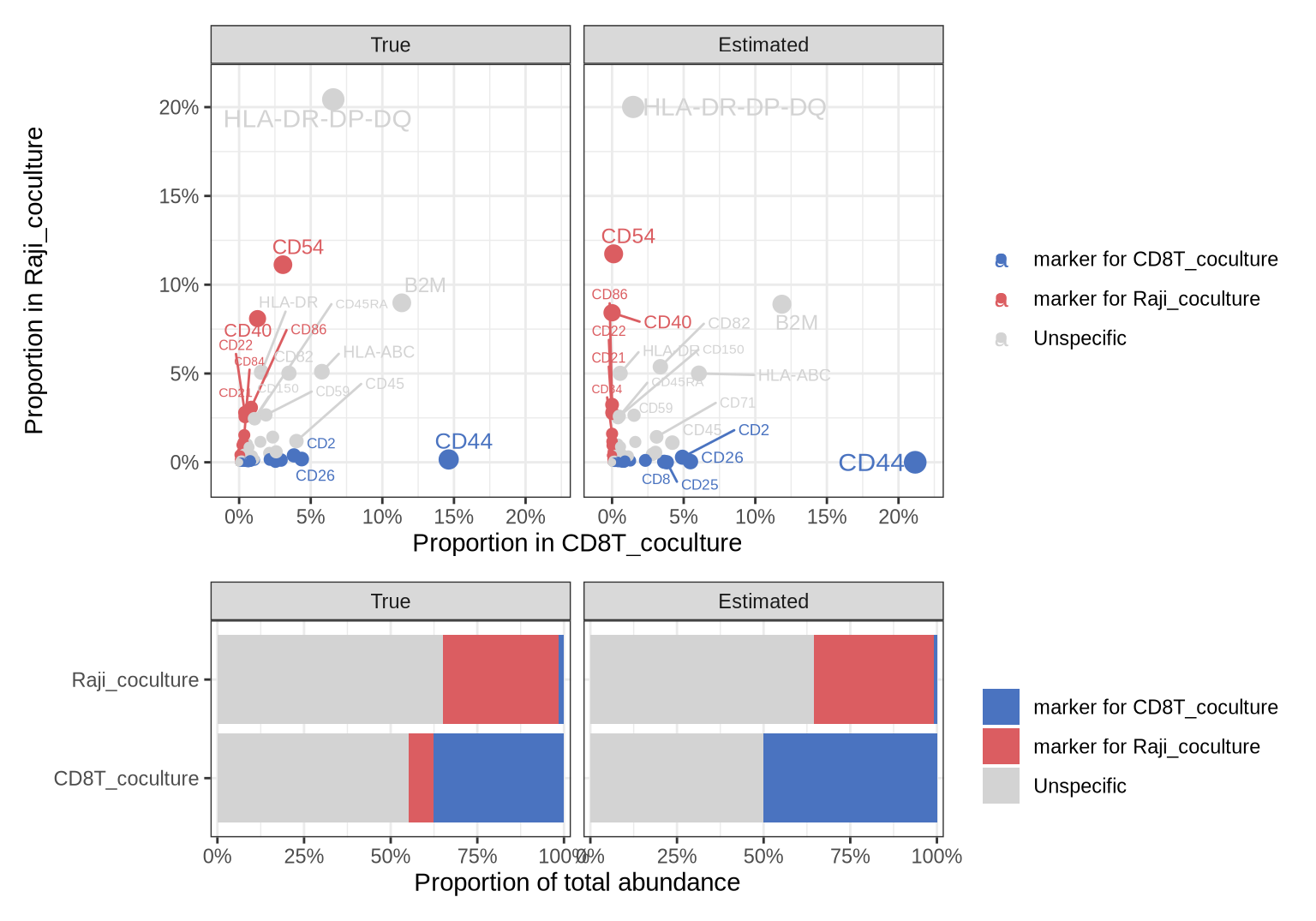
markers_df %>% head()
# A tibble: 6 × 6
marker target_unmixed_freq receiver_unmixed_freq target_freq receiver_freq
<chr> <dbl> <dbl> <dbl> <dbl>
1 HLA-ABC 0.0500 0.0606 0.0510 0.0578
2 B2M 0.0890 0.119 0.0898 0.114
3 CD11b 0.0000397 0.0000763 0.0000437 0.0000797
4 CD11c 0.0000243 0.000740 0.0000542 0.000629
5 CD18 0.000873 0.0127 0.00121 0.0106
6 CD82 0.0538 0.0337 0.0502 0.0348
# ℹ 1 more variable: label <glue>
The function returns a data frame with the protein IDs (“marker”), the true abundance proportions in the receiver/target populations and the estimated (unmixed) proportions in the receiver/target populations. The last column (“label”) specifies whether the marker is specific to the receiver population, target population or if it’s unspecific. The function also returns a plot summarizing the results.
In the top panel of the plot above we see the selected Raji-specific (target) markers in red, the CD8T-specific (receiver) markers in blue and the unspecific markers in grey. The left plot shows the true proportions in the co-culture data and the right plot shows the unmixed proportions. Here, we can for example see that CD54 looks quite abundant in the CD8T cells; however, after unmixing it becomes clear that CD54 is highly Raji-specific. Without unmixing, we would likely have ignored this marker which should to be an excellent candidate for patch detection since it is both highly abundant in and specific to the Raji cells.
The lower panel shows the total proportions for the selected markers in the two cell populations. Here, we see that the selected Raji protein markers make up roughly 10% of the total abundance in co-cultured CD8T cells, but virtually 0% after unmixing. Importantly, we want the proportion of Raji markers to be as high as possible in the Raji cells and as low as possible (preferably close to 0%) in the CD8T cells. This is crucial for efficient patch detection. As a rule of thumb, the total proportion of target marker proteins in the target population should be at least 20-30%.
Among the unspecific markers we find high abundant proteins such as HLA-ABC, B2M and CD150 that are present on both CD8 T cells and Raji cells. HLA-DR-DP-DQ is clearly more abundant on the Raji cells, but it is also present at relatively high levels on the CD8T cells, making it an unsuitable marker for patch detection.
Run patch detection
Now we are ready to proceed with the patch detection step. The method operates on the PNA cell graph, meaning that we first need to load these into our Seurat object. For this step, we will only need the CD8T cells from the co-culture experiment. The PNA cell graphs are large data structures and when loaded, they can consume a lot of memory. Although we only have 415 CD8 T cells from the co-culture experiment, they use up close to 8GB of memory. For larger datasets, it is generally a good idea to process smaller batches of cells.
# Select CD8T cell component IDs
cd8t_cells <- pg_data_combined[[]] %>%
filter(celltype_condition == "CD8T_coculture") %>% rownames()
# Load cell graphs
pg_data_combined <- LoadCellGraphs(pg_data_combined, cells = cd8t_cells, add_layouts = TRUE)
cg_list <- CellGraphs(pg_data_combined)[cd8t_cells]
pg_data_combined <- RemoveCellGraphs(pg_data_combined)
We have already selected the markers for the CD8T cells and the Raji cells, so we are now good to go. Markers for the receiver cells (CD8T) are optional but generally improves patch detection by blocking nodes in the graph that are receiver-cell specific.
You can find more information about the patch detection algorithm in the
function documentation (?patch_detection). The algorithm operates on
the PNA graphs and attempts to form subgraphs using an expand and
contract method initialized by nodes labelled with patch markers. This
is a computationally intensive step, so it may take a while to run.
# Select patch and receiver markers
receiver_markers <- markers_df %>%
filter(label == "marker for CD8T_coculture") %>%
pull(marker)
patch_markers <- markers_df %>%
filter(label == "marker for Raji_coculture") %>%
pull(marker)
# Run patch detection
cg_list <- pbapply::pblapply(cg_list, function(cg) {
cg <- suppressWarnings({
patch_detection(
cg,
# We only use the markers that are present in the counts matrix,
# otherwise the patch detection will fail.
patch_markers = intersect(patch_markers, colnames(cg@counts)),
receiver_markers = intersect(receiver_markers, colnames(cg@counts)),
# The smallest allowed patch size is set to 100
patch_nodes_threshold = 100,
verbose = FALSE
)
})
}, cl = NULL)
Inspect results
Let’s inspect the results in a single CellGraph object from cg_list.
Each CellGraph contains a tidy graph (visit
tidygraph for more information)
with a node table and an edge table. After running patch detection, the
node table will contain a patch column which indicates whether the
node is part of a patch or not. By convention, a value of 0 is used to
label receiver graph nodes, whereas the patches are labelled 1, 2, 3,
etc.
The node table also contains a column called potential_patch. These
are small patch subgraphs that were identified by the patch detection
algorithm but didn’t pass the size threshold (patch_nodes_threshold).
These are typically too small to evaluate downstream, but this
information can be useful to filter the receiver graph later.
One important point here is that each patch subgraph is guaranteed to be
connected, but the receiver subgraph is not. So if you were to segment
out the receiver subgraph from the CellGraph object, you’d likely end
up with a disconnected graph.
cg <- cg_list[[2]]
# Inspect the node table
CellGraphData(cg, "cellgraph") %N>%
as_tibble()
# A tibble: 94,892 × 4
name node_type patch potential_patch
<chr> <chr> <int> <int>
1 71905342332125-umi1 umi1 0 NA
2 22437986569621294-umi2 umi2 0 NA
3 116896337584537-umi1 umi1 0 NA
4 41362921692680373-umi2 umi2 0 NA
5 300756512162453-umi1 umi1 0 NA
6 57488365265918230-umi2 umi2 0 NA
7 376227976534695-umi1 umi1 0 NA
8 54716076737357242-umi2 umi2 3 NA
9 394167817161303-umi1 umi1 0 NA
10 56628804997723423-umi2 umi2 0 NA
# ℹ 94,882 more rows
# Count patch sizes
patch_sizes <- CellGraphData(cg, "cellgraph") %N>%
pull(patch) %>%
table()
glue::glue("Receiver subgraph size: {patch_sizes['0']}")
Receiver subgraph size: 85444
glue::glue("Patch subgraph sizes: {patch_sizes[setdiff(names(patch_sizes), '0')] %>% paste(collapse = ', ')}")
Patch subgraph sizes: 5912, 2474, 919, 143
Calculate patch sizes
From our list of CellGraphs, we can now calculate the patch sizes and
visualize the size distribution. We will also label patches as “large”
if they make up at least 1% of the total PNA graph size, “small”
otherwise.
patch_sizes <- lapply(names(cg_list), function(nm) {
cg <- cg_list[[nm]]
cg@cellgraph %N>%
pull(patch) %>%
table() %>%
enframe(name = "patch", value = "count") %>%
mutate(count = as.integer(count), component = nm)
}) %>% bind_rows()
patch_sizes <- patch_sizes %>%
left_join(pg_data_combined[[]] %>% select(n_umi) %>% as_tibble(rownames = "component"), by = "component") %>%
mutate(p = count / n_umi) %>%
mutate(type = case_when(p >= 0.01 ~ "large", TRUE ~ "small"))
patch_sizes %>% filter(patch > 0) %>%
ggplot(aes(count, fill = type)) +
geom_histogram(bins = 30, color = "black", position = "dodge") +
scale_x_log10() +
theme_bw() +
labs(x = "Patch size (UMI counts, log10)", y = "Number of patches")
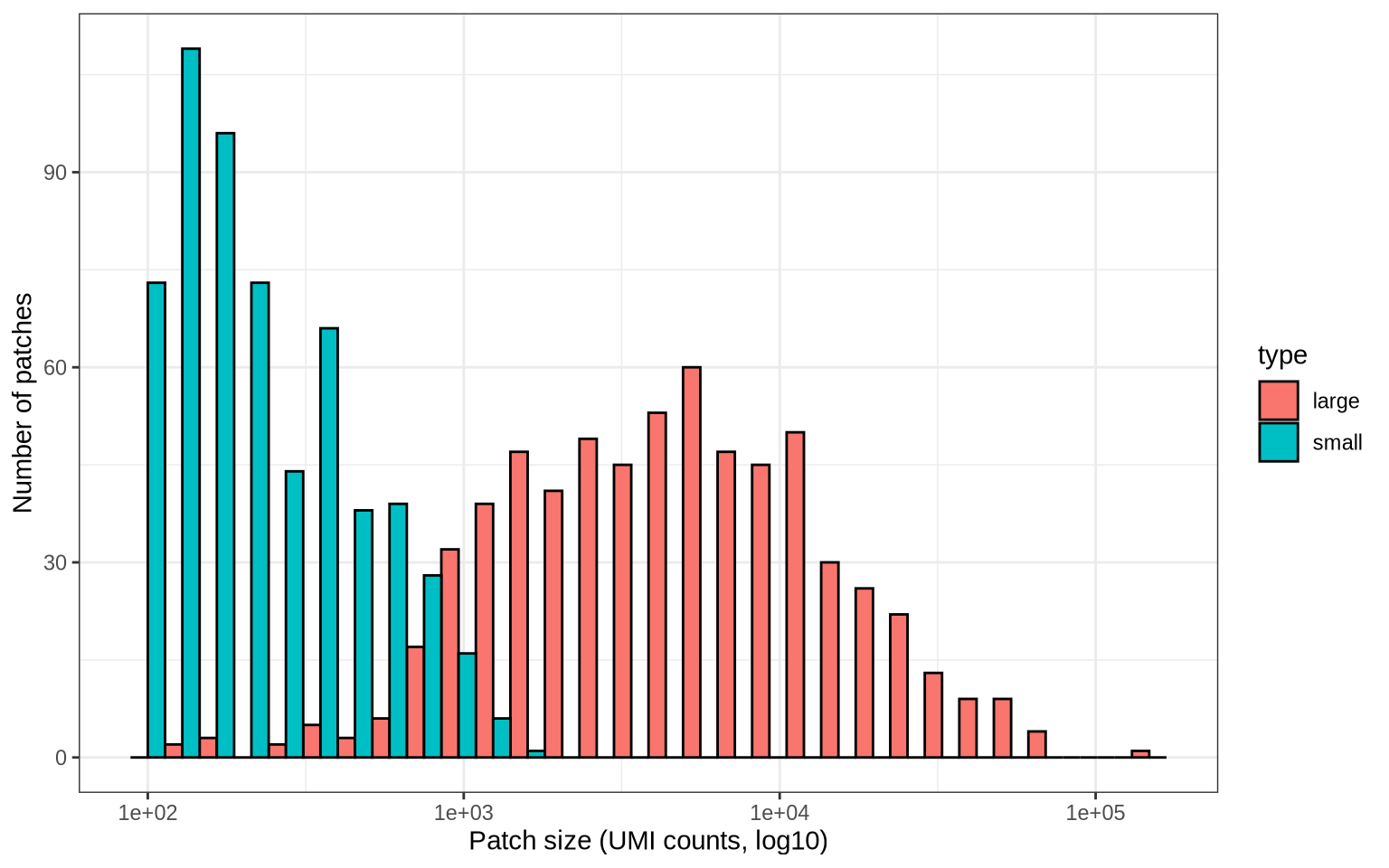
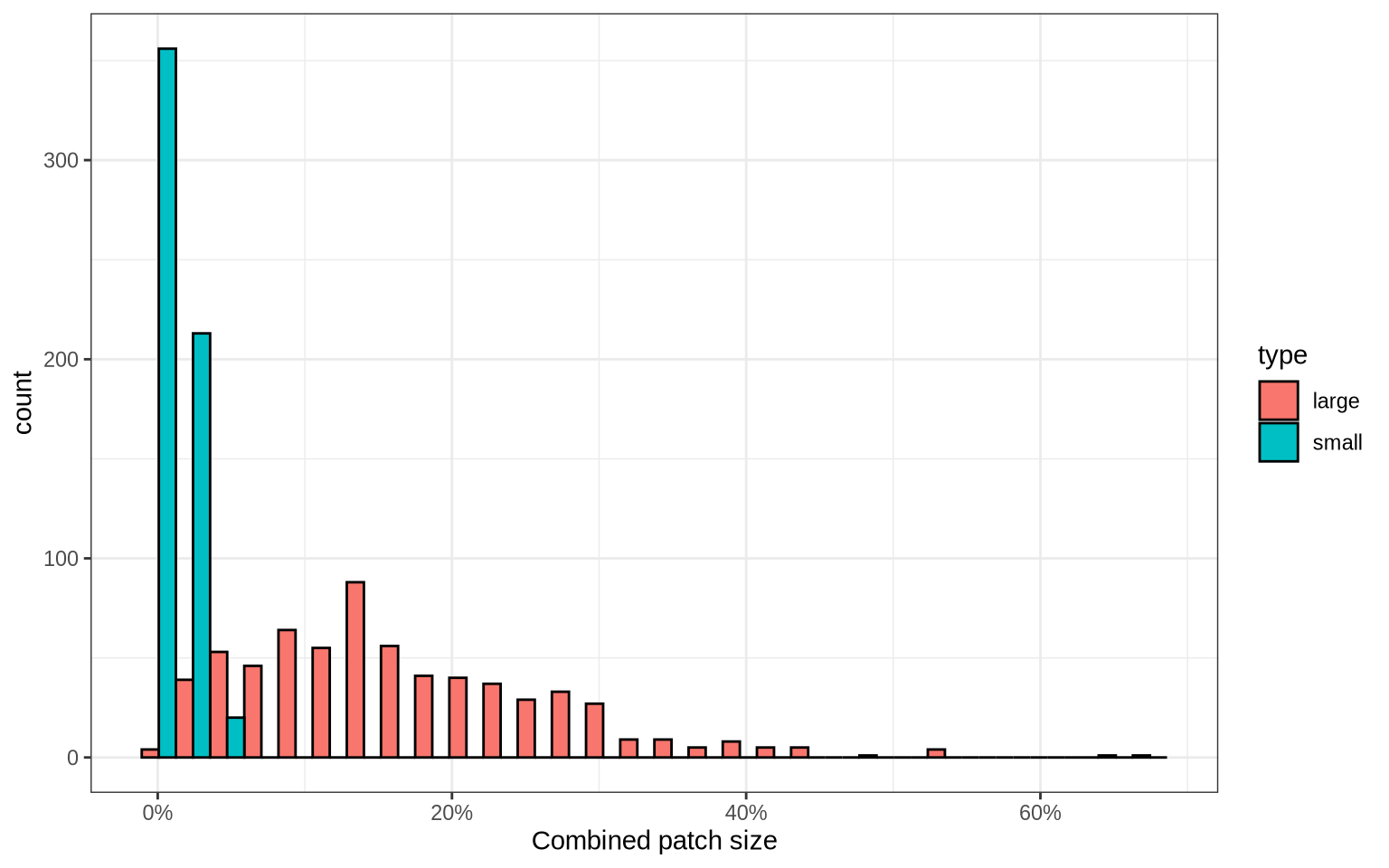
We can also visualize the proportion of nodes in the CD8T cell graphs that are labelled as patches. Roughly ~5-30% of nodes in CD8T cell graphs are found in the detected Raji patches.
patch_sizes %>%
filter(patch > 0) %>%
group_by(component, type) %>%
mutate(p = sum(p)) %>%
ggplot(aes(p, fill = type)) +
geom_histogram(bins = 30, color = "black", position = "dodge") +
theme_bw() +
scale_x_continuous(labels = scales::percent) +
labs(x = "Combined patch size")
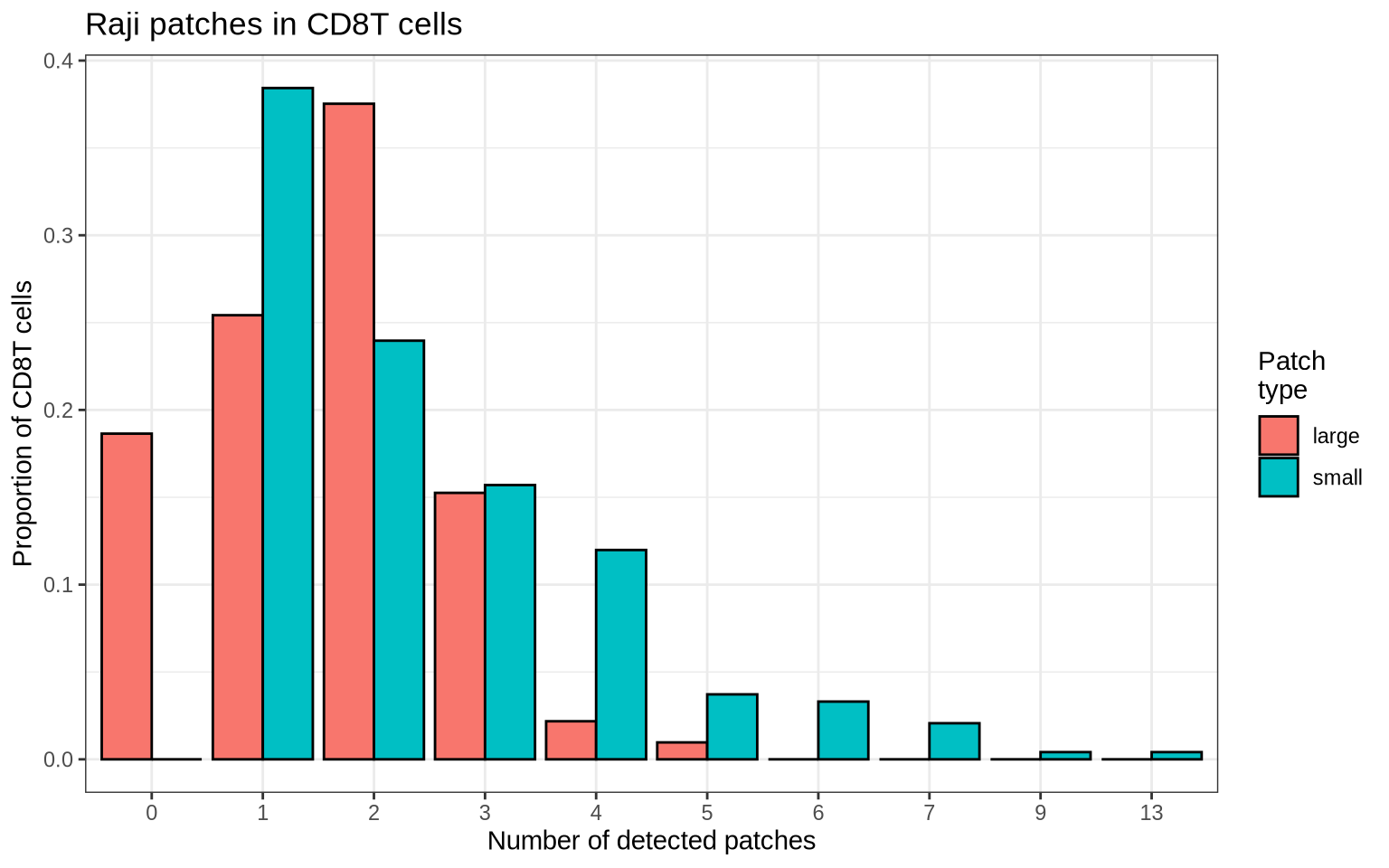
Finally, let’s count the number of patches. Here, we see that most CD8T cells have 0-3 large patches and often several smaller patches.
patch_sizes_summarized <- patch_sizes %>%
group_by(component, type) %>%
count() %>%
mutate(n = if_else(type == "large", n - 1, n))
patch_sizes_summarized %>%
group_by(type, n) %>%
count(name = "count") %>%
ungroup() %>%
complete(type, n, fill = list(count = 0)) %>%
group_by(type) %>%
mutate(count = count / sum(count)) %>%
mutate(n = factor(n)) %>%
ggplot(aes(n, count, fill = type)) +
geom_col(position = "dodge", color = "black") +
theme_bw() +
labs(y = "Proportion of CD8T cells",
x = "Number of detected patches",
title = "Raji patches in CD8T cells",
fill = "Patch\ntype")
Patch protein composition
Now the question is, how reliable are these patches? To answer this, we can look at the protein composition of the patches. We anticipate that patches will be enriched for Raji protein markers whereas the receiever subgraphs should be enriched for CD8T cell protein markers. We will collapse the counts matrices from the graphs to patch level and then visualize the protein composition of the patches. This step can take several minutes to complete.
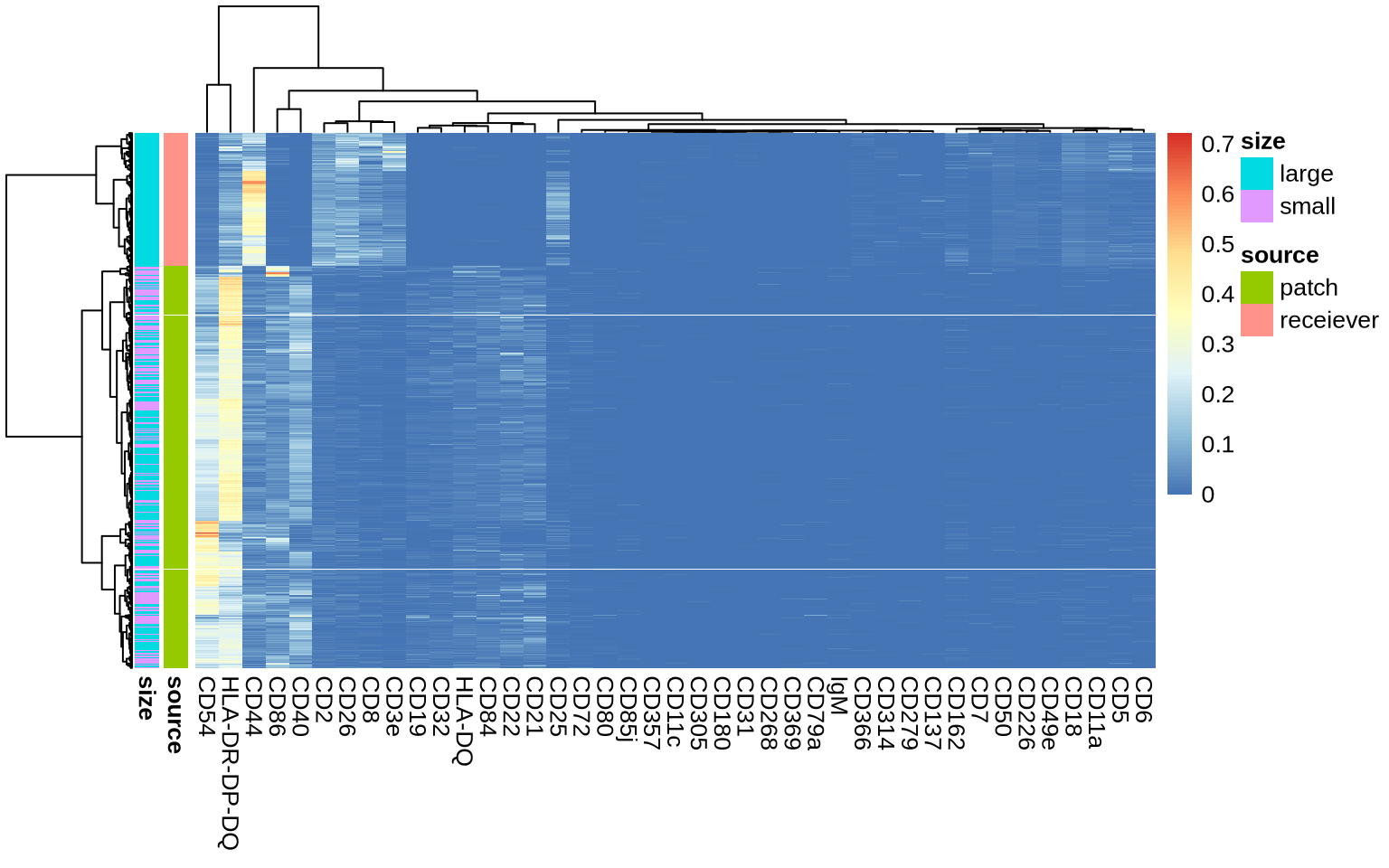
As the heatmap below demonstrates, our expectations are confirmed. Nevertheless, it’s crucial to acknowledge the inherent complexity of patch detection, which can lead to a certain degree of “bleedover” or signal contamination between the segmented receiever and patch subgraphs.
collapsed_counts <- pbapply::pblapply(names(cg_list), function(nm) {
cg <- cg_list[[nm]]
tibble(patch = cg@cellgraph %>% pull(patch) %>% as.factor(),
potential_patch = cg@cellgraph %>% pull(potential_patch)) %>%
bind_cols(as.matrix(cg@counts)) %>%
filter(is.na(potential_patch) | patch != 0) %>%
group_by(patch) %>%
summarize(across(where(is.numeric), ~ sum(.x))) %>%
mutate(component = nm)
}) %>% bind_rows()
collapsed_matrix <- collapsed_counts %>%
mutate(id = paste0(component, "-", patch)) %>%
select(-patch, -component) %>%
data.frame(row.names = "id", check.names = FALSE) %>%
as.matrix()
collapsed_matrix[is.na(collapsed_matrix)] <- 0
ann <- data.frame(
source = if_else(collapsed_counts$patch == 0, "receiever", "patch"),
size = patch_sizes$type,
row.names = rownames(collapsed_matrix)
)
collapsed_matrix[, c(patch_markers, receiver_markers, "HLA-DR-DP-DQ")] %>%
prop.table(margin = 1) %>%
pheatmap::pheatmap(annotation_row = ann, show_rownames = FALSE, clustering_method = "ward.D2")
Re-analysis of segmented data
With our new count matrix representing segmented receiver graphs and patches, we can now create a Seurat object to plug into a data-driven analysis workflow. Let’s create this object and add some relevant meta data. We can also examine the size (UMI count) distribution to compare the patch sizes with the receiever graph sizes.
pg_data_segmented <- CreateSeuratObject(counts = t(collapsed_matrix), assay = "segmented")
pg_data_segmented$n_umi <- Matrix::colSums(LayerData(pg_data_segmented, layer = "counts"))
pg_data_segmented$type <- ifelse(collapsed_counts$patch == 0, "receiever", "patch")
pg_data_segmented$original_component <- stringr::str_remove(colnames(pg_data_segmented), "-[0-9]+$")
MoleculeRankPlot(pg_data_segmented, group_by = "type")
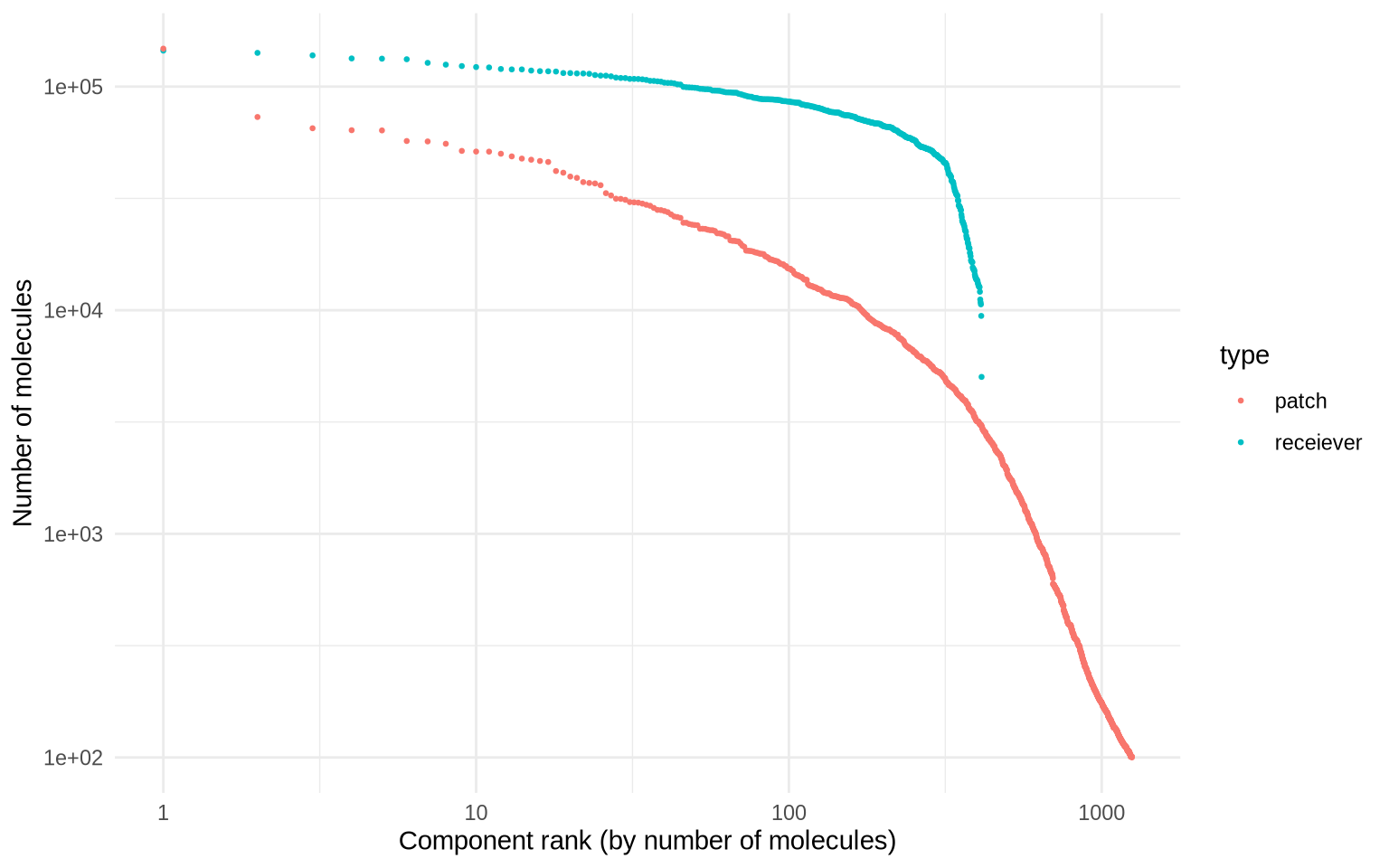
As we already know, a large fraction of the detected patches are very small. For the re-analysis, we will only keep patches that have more than 2000 UMI counts to ensure that we have sufficient data per graph component to work with.
# Subset data to include graphs with more than 2000 UMI counts
pg_data_segmented <- pg_data_segmented %>% subset(n_umi > 2e3)
VariableFeatures(pg_data_segmented) <- rownames(pg_data_segmented)
pg_data_segmented <- pg_data_segmented %>%
Normalize(method = "clr") %>%
ScaleData() %>%
RunPCA() %>%
RunUMAP(dims = 1:6, reduction = "pca") %>%
FindNeighbors(dims = 1:6, reduction = "pca") %>%
FindClusters(resolution = 0.2, verbose = FALSE)
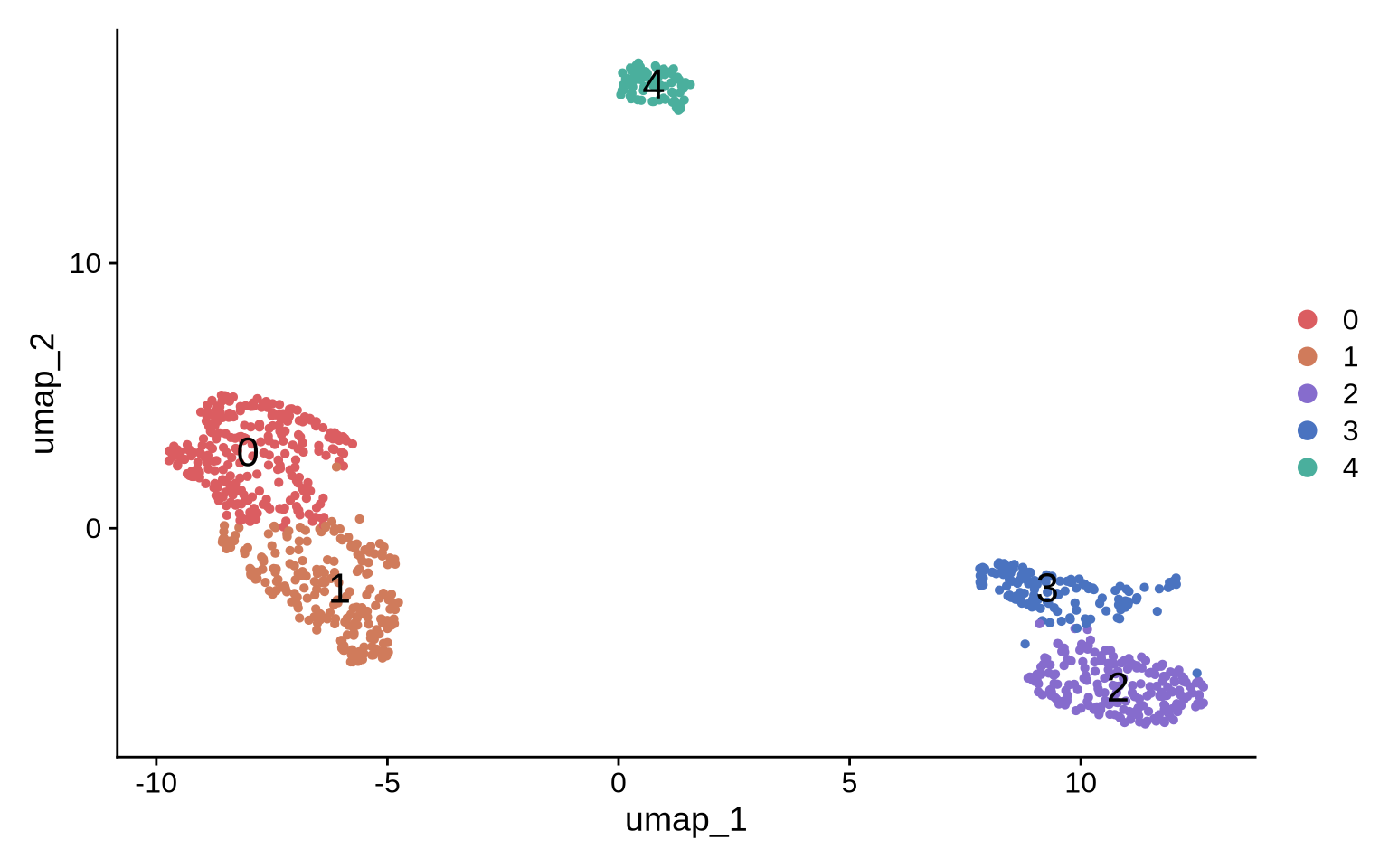
DimPlot(pg_data_segmented, label = TRUE, label.size = 6, cols = plot_colors[c(6:7, 1:3)])
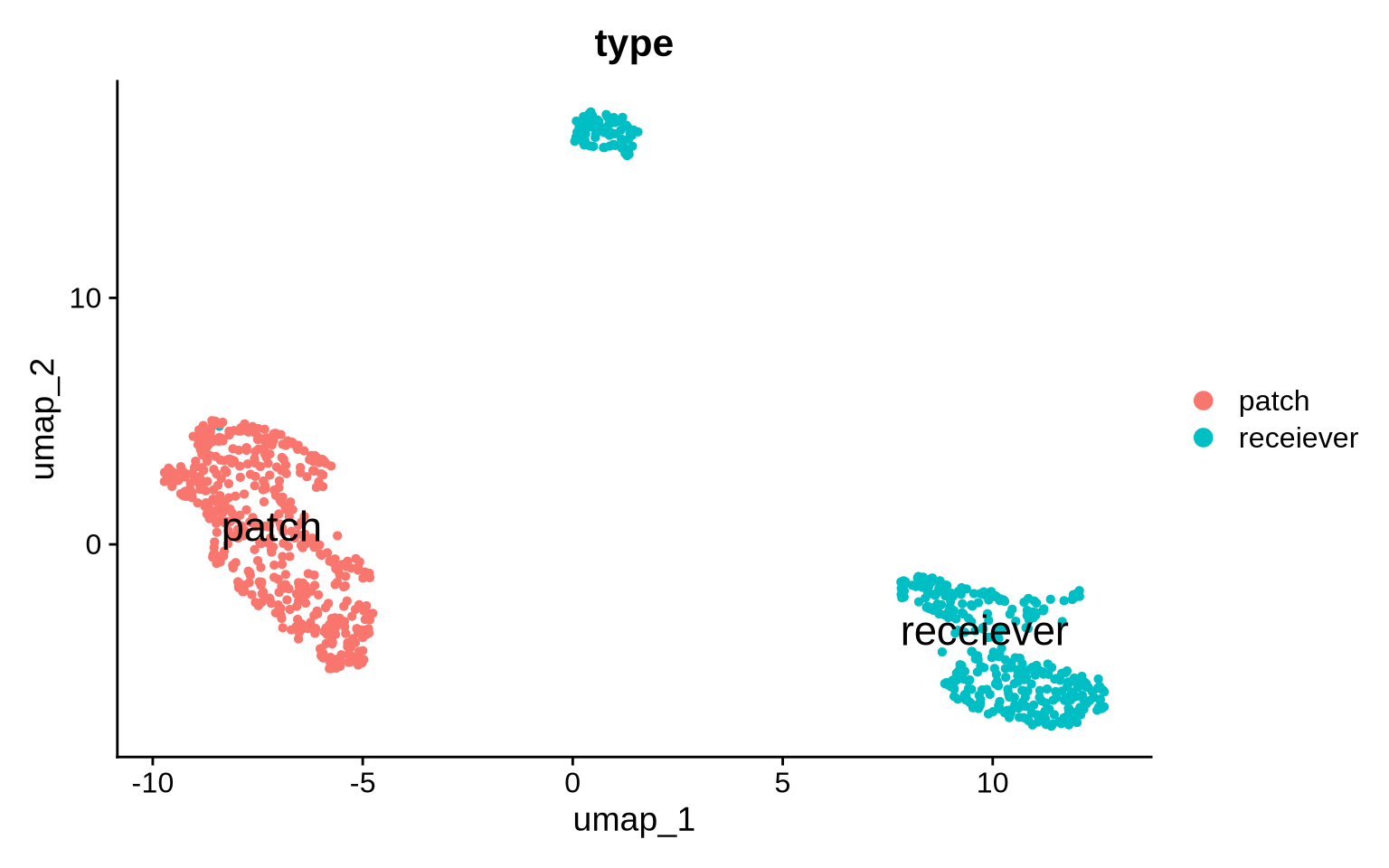
DimPlot(pg_data_segmented, label = TRUE, label.size = 6, group.by = "type")
Compare segmented cells with original CD8T cells
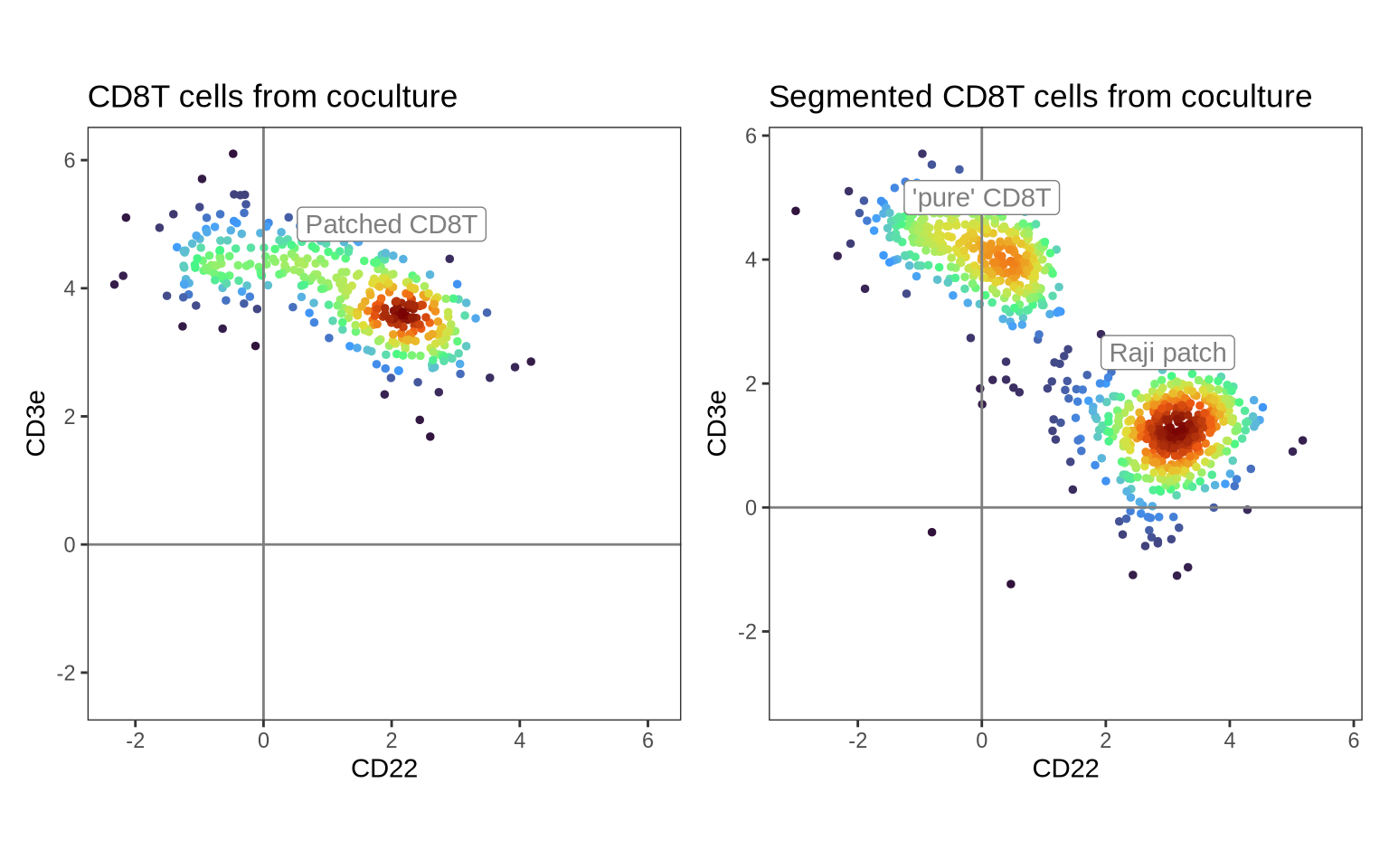
Let’s compare our segmented data with the original CD8T cells from the co-culture. In the left panel, we see the original CD8T cell population enriched for the Raji protein marker CD22. After segmenting this population, the patched CD8T cells are split into two populations: “pure” CD8T cells and Raji patches.
p1 <- DensityScatterPlot(pg_data_combined %>% subset(condition == "coculture") %>% subset(celltype == "CD8T"),
marker1 = "CD22", marker2 = "CD3e", margin_density = FALSE) &
geom_label(data = tibble(
marker1 = 2,
marker2 = 5,
label = "Patched CD8T",
dens = NA_real_
), aes(marker1, marker2, label = label)) &
ggtitle("CD8T cells from coculture") &
guides(color = "none")
p2 <- DensityScatterPlot(pg_data_segmented, marker1 = "CD22", marker2 = "CD3e", margin_density = FALSE) &
geom_label(data = tibble(
marker1 = c(0, 3),
marker2 = c(5, 2.5),
label = c("'pure' CD8T", "Raji patch"),
dens = NA_real_
), aes(marker1, marker2, label = label)) &
ggtitle("Segmented CD8T cells from coculture") &
guides(color = "none")
p1 + p2
Profile and annotate segmented data
Now we can go ahead and identify and visualize marker proteins for each cluster. We find two Raji patch populations (clusters 0 and 1) and three CD8T populations (clusters 2, 3 and 4).
de_markers <- FindAllMarkers(pg_data_segmented %>% JoinLayers(), fc.name = "difference", mean.fxn = Matrix::rowMeans, only.pos = TRUE)
sel_markers <- de_markers %>%
filter(pmax(pct.1, pct.2) > 0.2 & p_val_adj < 0.01) %>%
group_by(cluster) %>%
arrange(-difference) %>%
slice_head(n = 20) %>%
pull(gene) %>%
unique()
pg_data_segmented$seurat_clusters <- pg_data_segmented$seurat_clusters %>% factor(levels = c("0", "1", "2", "3", "4")) %>% unname()
pg_data_segmented <- SetIdent(pg_data_segmented, value = "seurat_clusters")
DoHeatmap(pg_data_segmented, features = sel_markers, group.colors = plot_colors[c(6:7, 1:3)])
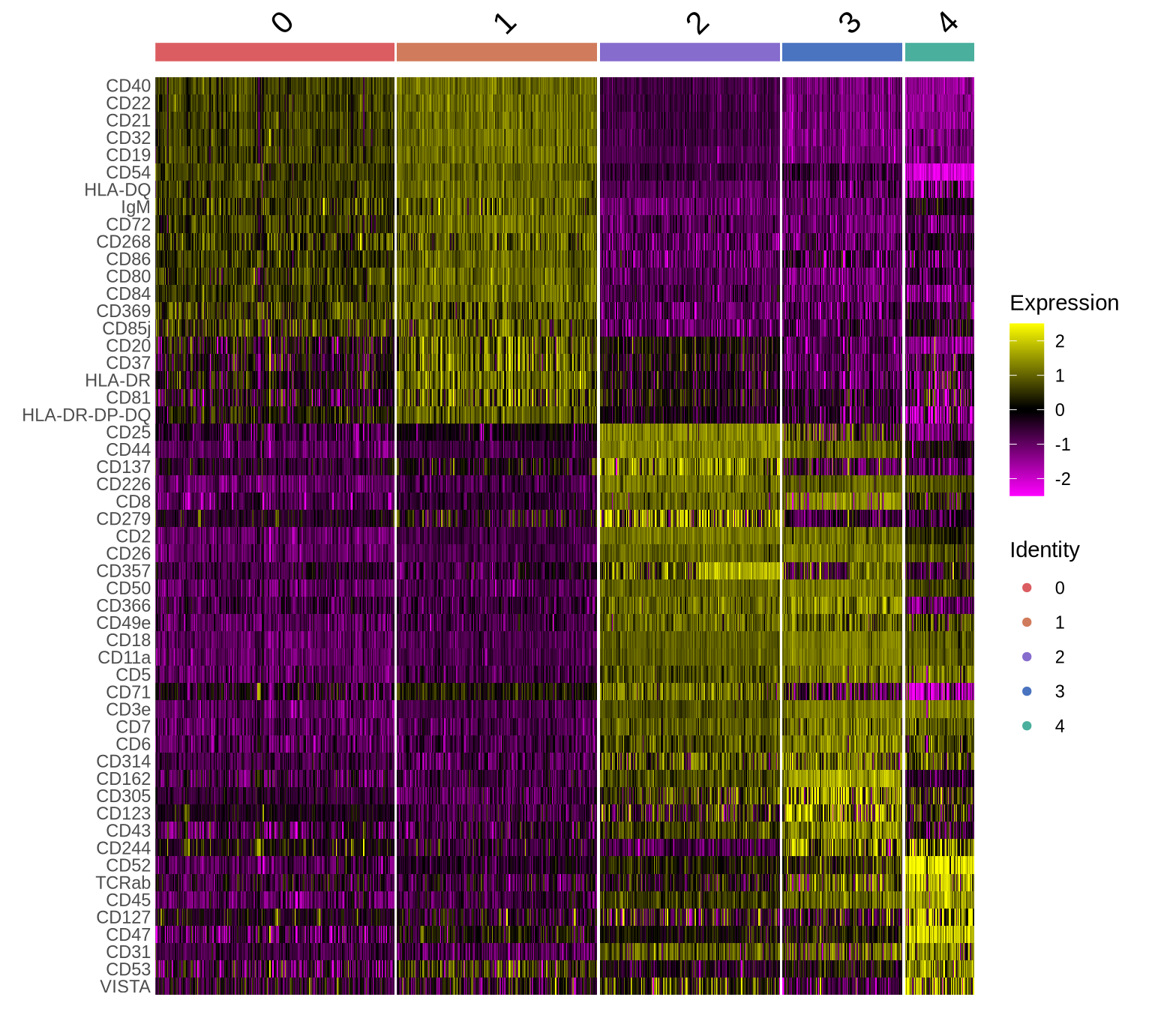
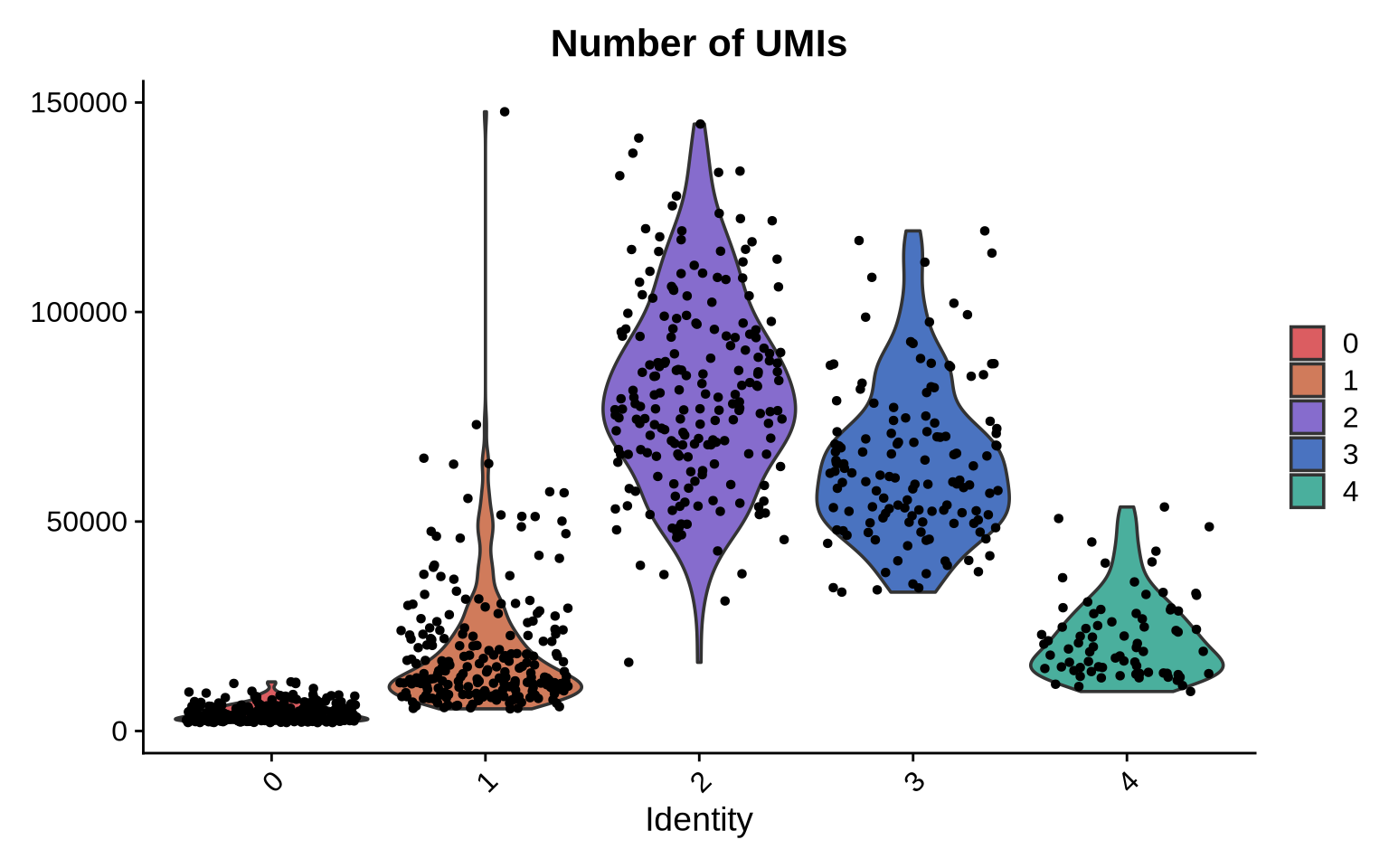
At a first glance, it looks like we have two Raji patch populations; however, this separation is likely driven by the difference in sizes. The patches in cluster 0 are much smaller which is likely why some Raji markers are missing, thereby driving the separation of these two clusters.
VlnPlot(pg_data_segmented, feature = "n_umi", cols = plot_colors[c(6:7, 1:3)]) &
ggtitle("Number of UMIs")
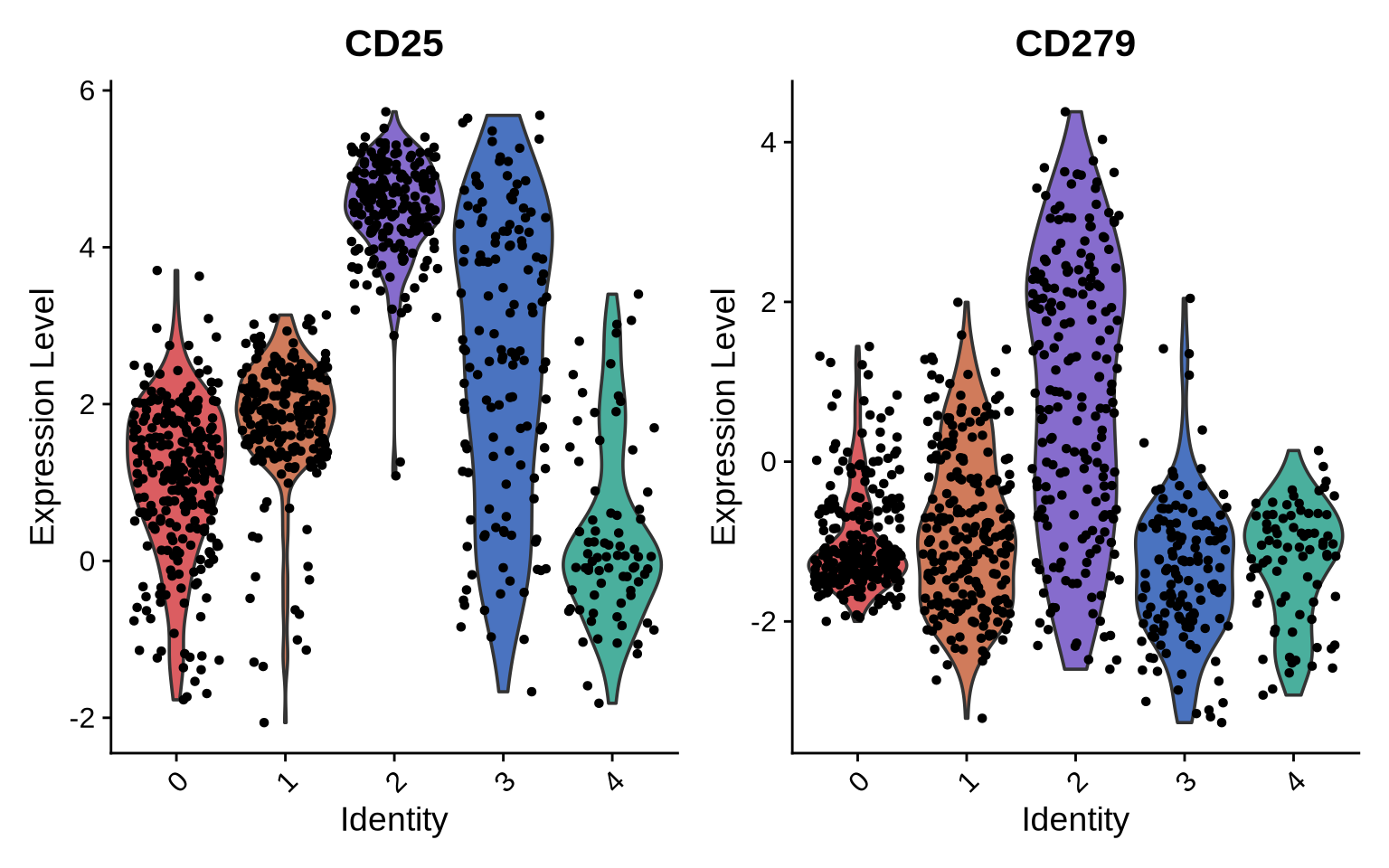
The CD8T cell populations can be classified into three different activation states based on the abundance of CD25 and CD279:
-
non-activated: CD25-/CD279-
-
activated: CD25+/CD279-
-
exhausted: CD25+/CD279+
VlnPlot(pg_data_segmented, features = c("CD25", "CD279"), cols = plot_colors[c(6:7, 1:3)])
Now we can annotate the clusters accordingly:
ann <- c(
"0" = "Raji patch",
"1" = "Raji patch",
"2" = "CD8T exhausted",
"3" = "CD8T activated",
"4" = "CD8T non-activated"
)
pg_data_segmented$celltype <- ann[as.character(pg_data_segmented$seurat_clusters)] %>% unname() %>%
factor(levels = c("Raji patch",
"CD8T non-activated",
"CD8T activated",
"CD8T exhausted"))
cols <- c("Raji patch" = plot_colors[6],
"CD8T non-activated" = plot_colors[1],
"CD8T activated" = plot_colors[2],
"CD8T exhausted" = plot_colors[3])
DimPlot(pg_data_segmented, label = TRUE, label.size = 4, group.by = "celltype", cols = cols)
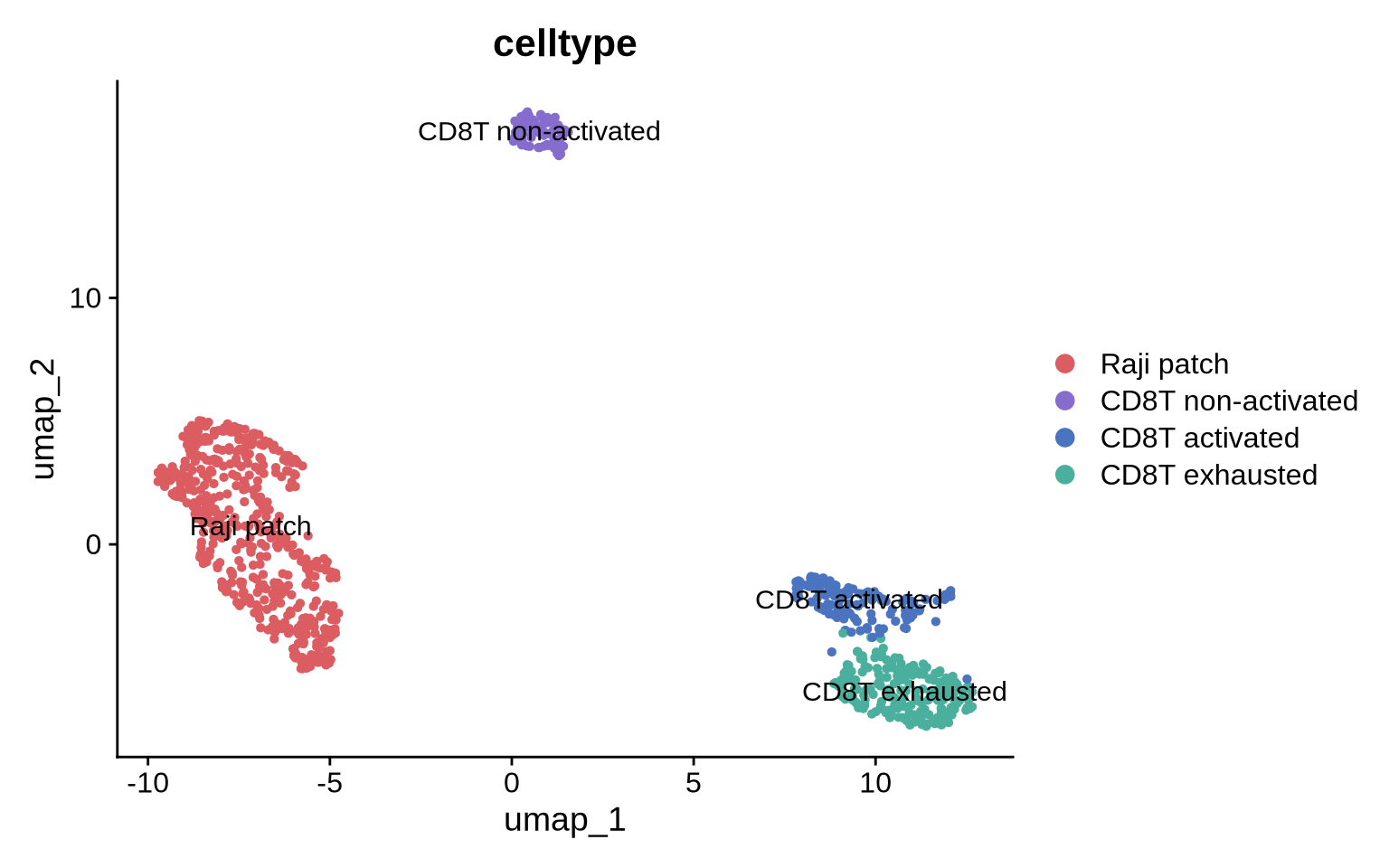
T cell states and patches
Due to the relative homogeneity of the Raji population, there’s currently no data indicating an association between distinct Raji phenotypes and the three CD8T cell states. Nevertheless, it’s possible to examine the connection between CD8T cell states and two key parameters: the count of Raji patches and their aggregate size. To begin, we will subset our segmented CD8T cells and add data on patch quantity.
pg_data_cd8t <- pg_data_segmented %>%
subset(celltype %in% c("CD8T non-activated", "CD8T activated", "CD8T exhausted"))
pg_data_cd8t$n_large_patches <- pg_data_cd8t[[]] %>%
left_join(patch_sizes_summarized %>% filter(type == "large"), by = c("original_component" = "component")) %>%
mutate(n = if_else(is.na(n), 0, n)) %>%
pull(n)
pg_data_cd8t$n_small_patches <- pg_data_cd8t[[]] %>%
left_join(patch_sizes_summarized %>% filter(type == "small"), by = c("original_component" = "component")) %>%
mutate(n = if_else(is.na(n), 0, n)) %>%
pull(n)
patch_p_summarized <- patch_sizes %>%
filter(patch > 0) %>% # Exclude receiver
group_by(component, type) %>%
summarize(p = sum(p), .groups = "drop")
pg_data_cd8t$p_large_patches <- pg_data_cd8t[[]] %>%
left_join(patch_p_summarized %>% filter(type == "large"), by = c("original_component" = "component")) %>%
mutate(p = if_else(is.na(p), 0, p)) %>%
pull(p)
pg_data_cd8t$p_small_patches <- pg_data_cd8t[[]] %>%
left_join(patch_p_summarized %>% filter(type == "small"), by = c("original_component" = "component")) %>%
mutate(p = if_else(is.na(p), 0, p)) %>%
pull(p)
pg_data_cd8t$n_patches <- pg_data_cd8t$n_large_patches + pg_data_cd8t$n_small_patches
pg_data_cd8t$p_patches <- pg_data_cd8t$p_large_patches + pg_data_cd8t$p_small_patches
cd8t_patch_data <- FetchData(pg_data_cd8t, vars = c("p_patches", "n_patches", "celltype"))
p1 <- cd8t_patch_data %>%
ggplot(aes(celltype, n_patches, fill = celltype)) +
geom_violin(draw_quantiles = 0.5, scale = "width") +
labs(x = "", y = "Number of detected Raji patches")
p2 <- cd8t_patch_data %>%
ggplot(aes(celltype, p_patches, fill = celltype)) +
geom_violin(draw_quantiles = 0.5, scale = "width") +
scale_y_continuous(labels = scales::percent) +
labs(x = "", y = "Percent of graph covered by Raji patches")
p1 + p2 & theme_bw() &
scale_fill_manual(values = cols) &
theme(axis.text.x = element_text(angle = 45, hjust = 1),
legend.position = "none")
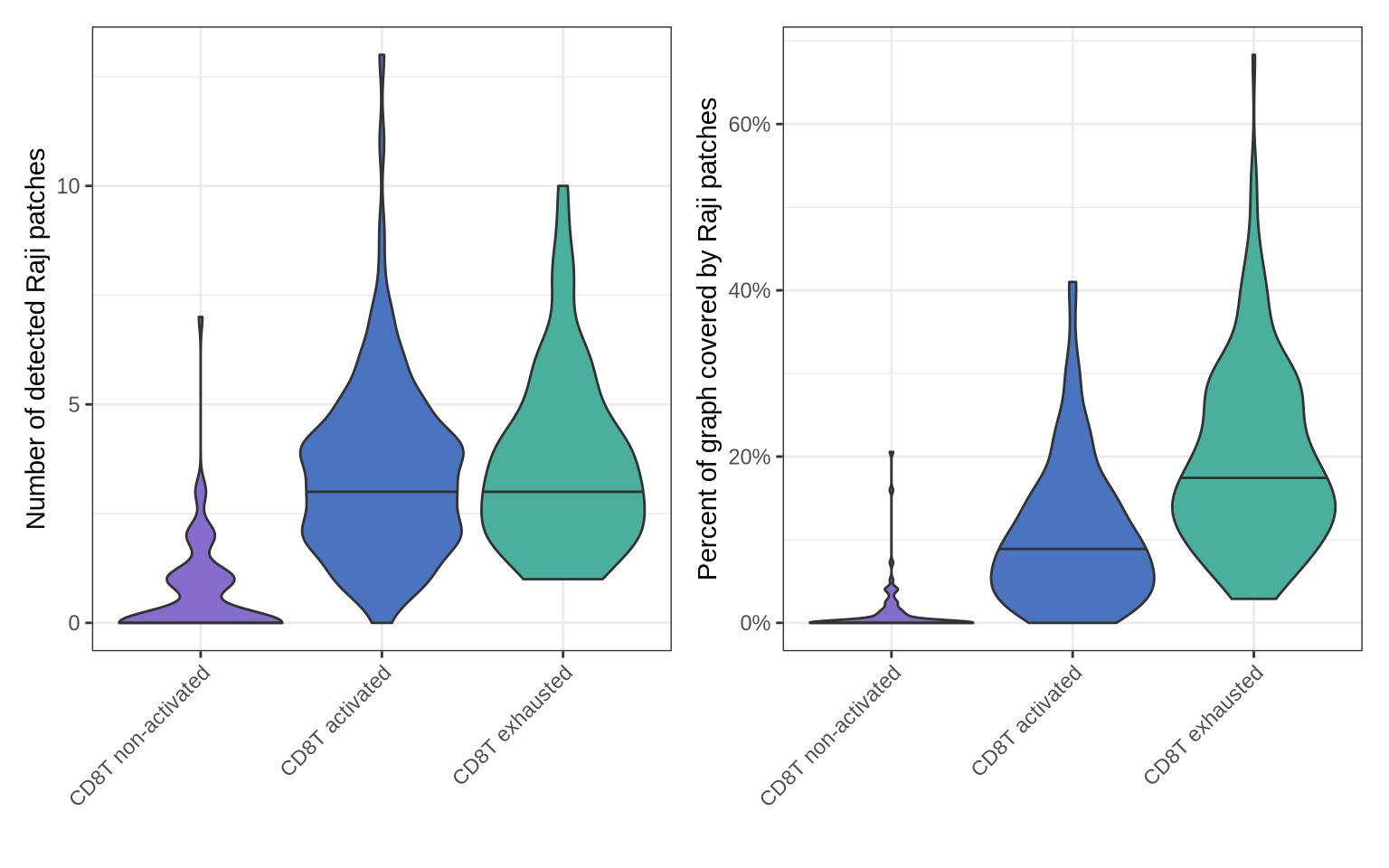
These plots show that activated CD8 T cells form more Raji patches and exhibit greater Raji patch coverage than non-activated cells. Exhausted CD8 T cells display even higher numbers of patches and more extensive Raji patch coverage compared to activated cells.
The increased patch formation on activated CD8 T cells compared to non-activated ones reflects normal immune engagement. It’s expected that activated CD8 T cells would interact more extensively with Raji cells than non-activated (naïve or resting memory) T cells. Activation primes CD8 T cells to recognize and engage with target cells, so an increased number of patches and patch coverage reflect this functional engagement.
The further increase in patch formation and coverage on exhausted CD8 T cells, when linked with the knowledge that chronic antigen exposure drives exhaustion, suggests that these exhausted cells have a history of, or are potentially trapped in, a state of persistent interaction with the antigen-presenting Raji cells. This extensive physical contact, which initially was part of an active immune response, becomes a contributing factor to their eventual exhaustion when sustained over time. The patch data provides a potential correlate for the intense and prolonged stimulation that defines the pathway to T cell exhaustion.
Visualize patched cells
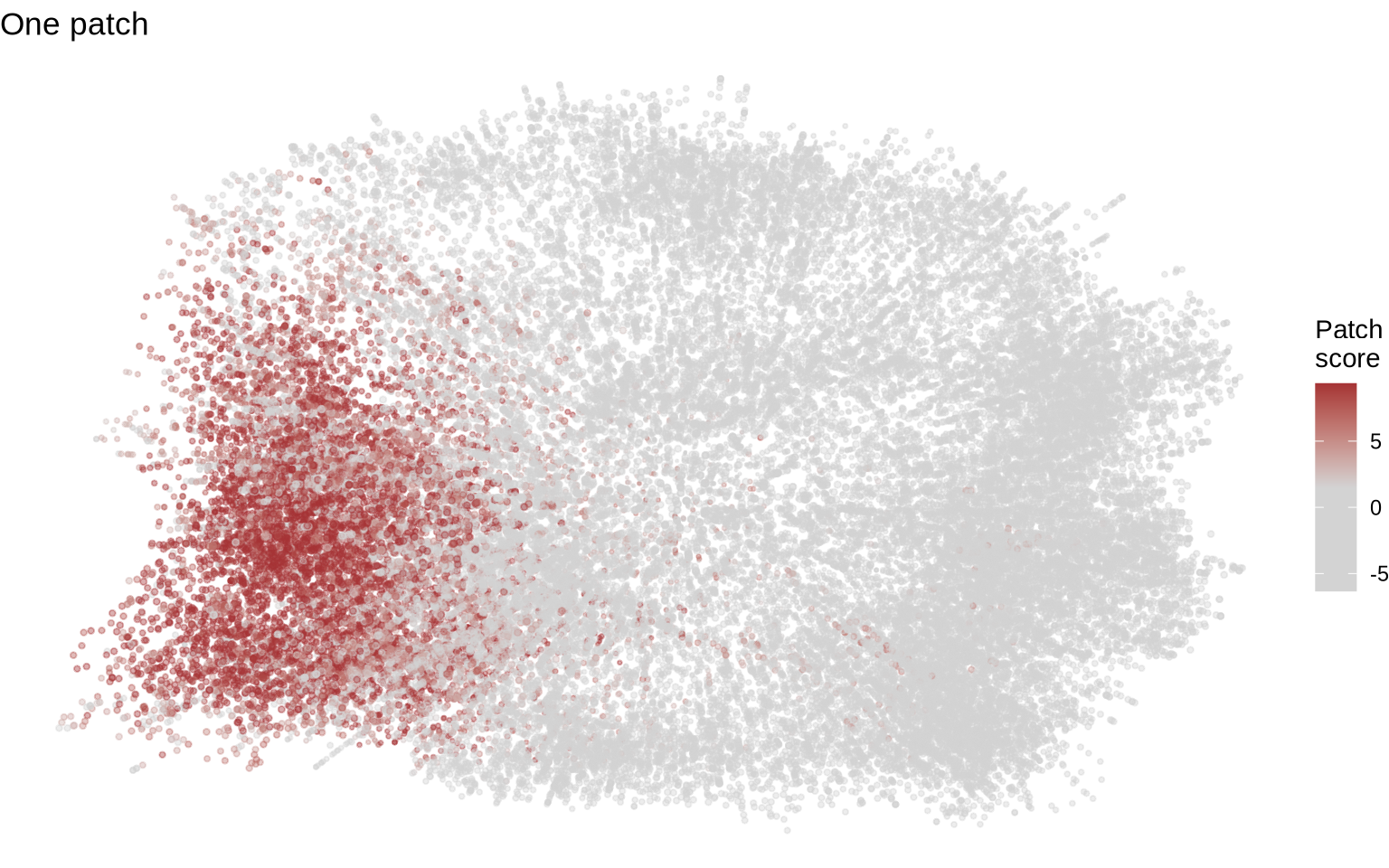
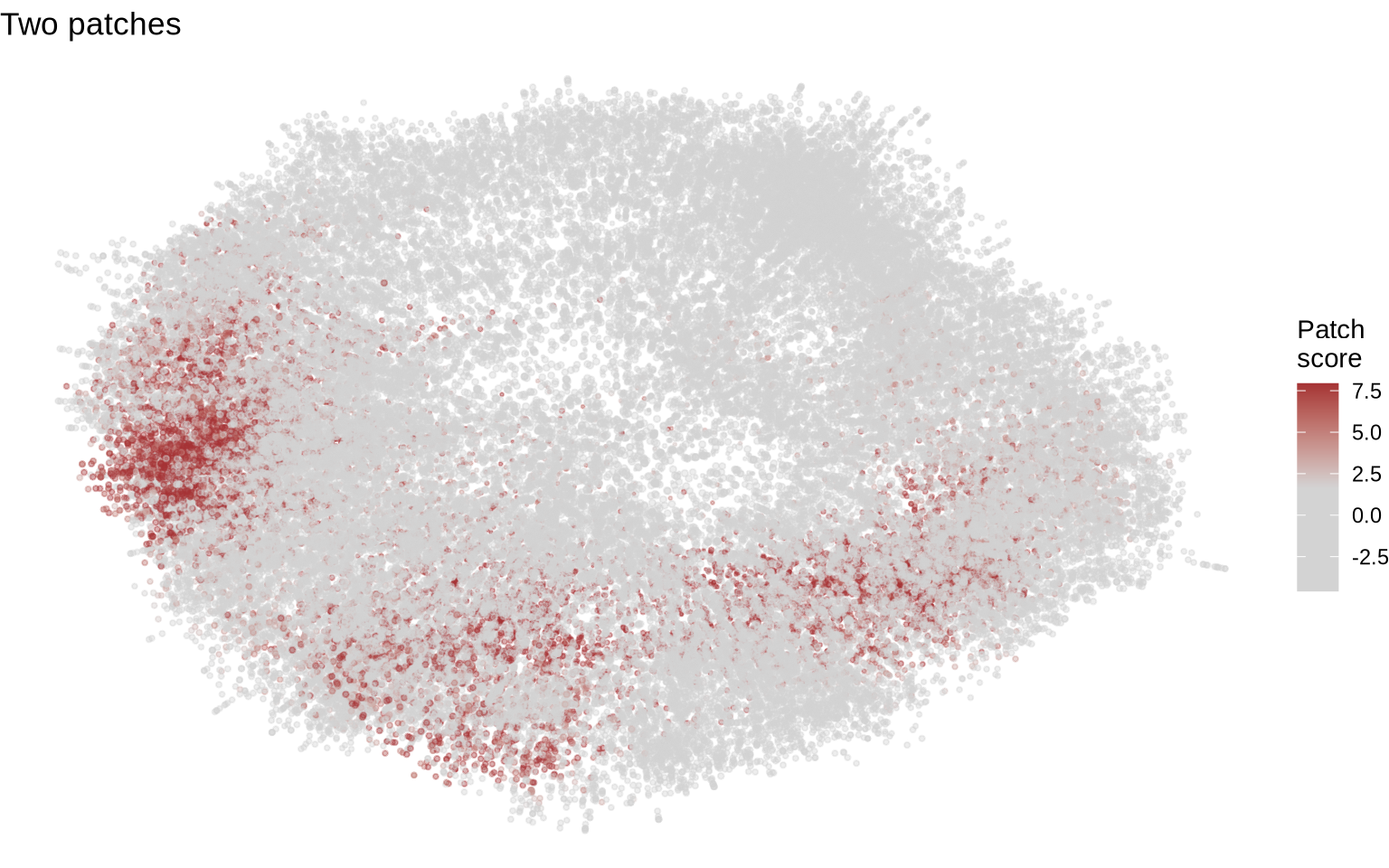
Visualizing patches on CD8T cell graphs can be difficult. The primary issue is that patches vary widely in size and can adopt irregular shapes within the graph layouts, making straightforward visualization challenging. Furthermore, layouts containing patches are frequently more distorted than those without, and the 3D nature of these layouts makes it even harder to view every patch simultaneously. Remember that the PNA graph layouts are abstract representations of the cells and are influenced by a number of factors, in particular the local connectivity which is expected vary more in patched cells. With this in mind, you will probably need to manually inspect multiple layout to find good examples for visualization.
We’ll use our patch count table to find cells with different numbers of patches. We’ve chosen two specific cells from the table below: one with a single large patch and another with two large patches.
# Use table of patch sizes to filter for
# cells with a desired number of patches
patch_sizes_summarized %>%
group_by(component) %>%
summarize(n = sum(n))
# A tibble: 413 × 2
component n
<chr> <dbl>
1 coculture1_01aa1d5b53369b6a 0
2 coculture1_022a17d9821b0dc0 4
3 coculture1_024b268fc621415c 3
4 coculture1_02e7aba0141f3781 3
5 coculture1_05ac7678d6bbff1b 0
6 coculture1_07594a52dfe38946 4
7 coculture1_0795eec3516219ae 2
8 coculture1_07fc35489bef2d9a 4
9 coculture1_090c364b51643c5e 0
10 coculture1_096893e9a4bbcc1c 0
# ℹ 403 more rows
plot_patches <- function(cg) {
xyz <- cg@layout$wpmds_3d
gi_mat <- local_G(cg@cellgraph, counts = as(matrix((cg@cellgraph %N>% pull(patch)) - 1), "dgCMatrix"), use_weights = FALSE, k = 4)
xyz$g <- gi_mat[, 1, drop = TRUE] %>% pixelatorR:::.trim_quantiles(top_q = 0.9)
xyz %>%
arrange(z) %>%
ggplot(aes(x, y, color = g, size = z)) +
geom_point(alpha = 0.4) +
scale_size(range = c(0.01, 1)) +
theme_void() +
scale_color_gradientn(colors = c("lightgrey", "lightgrey", "#A53335")) +
guides(size = "none") +
labs(color = "Patch\nscore")
}
cg1 <- cg_list[["coculture1_2a77aaf817b6376e"]]
cg2 <- cg_list[["coculture2_1c18b49416fba393"]]
plot_patches(cg1) + ggtitle("One patch")
plot_patches(cg2) + ggtitle("Two patches")
In this tutorial, we gained a comprehensive understanding of patch analysis in PNA data. We learned how to:
-
Understand patches as connected subgraphs enriched for specific protein markers, representing entities like cell fragments or interacting cells.
-
Perform quality control, data integration (using Harmony for co-culture data), and initial cell type annotation.
-
Utilize pixelatorR’s specialized function to unmix abundance data and select optimal receiver and target population markers for robust patch detection.
-
Apply the patch detection algorithm on PNA cell graphs.
-
Calculate and visualize patch sizes, proportions of cell graph coverage, and the number of patches per cell, differentiating between “large” and “small” patches.
-
Verify the purity of detected patches by analyzing their protein composition, confirming enrichment for target markers and depletion of receiver markers.
-
Create a new Seurat object from segmented receiver graphs and patches, allowing for independent clustering and annotation of these distinct cellular components.
-
Explore the relationship between CD8 T cell activation states and the quantity/coverage of Raji patches, revealing potential biological insights into cell-cell interactions and T cell exhaustion.
-
Understand the challenges and strategies for visually representing patches within 3D cell graph layouts.
This tutorial has equipped you with the foundational knowledge and practical skills to perform patch analysis, moving beyond protein abundance to explore the spatial organization interacting cells.